Abstract
The present study aimed to develop a practical method for preparing nanosuspension formulations of poorly water-soluble compounds for enhancing oral absorption in toxicology studies in the discovery stage. To obtain a suitable nanosuspension formulation for the intended purpose, formulations were optimized with a focus on the following characteristics: i) containing a high drug concentration, ii) consisting of commonly used excipient types in proper quantities for toxicology studies, iii) having long-term stability, and iv) having versatility for use with diverse compounds. Test compounds were milled with various excipients by wet media milling methods using a mixer mill (10 mg/batch) and a rotation/revolution mixer (0.5 g/batch). As a result, 100 mg/mL nanosuspensions of all 11 test compounds could be prepared with an optimized dispersing agent, 0.5% hydroxypropyl methylcellulose (HPMC) (3 cP)–0.5% Tween 80. Notably, it was found that the molecular weight of HPMC influenced not only particle size but also the stability of nanosuspensions and they were stable for 4 weeks at 5°C. The nanosuspensions increased in vitro dissolution rates and provided 3.9 and 3.0 times higher Cmax and 4.4 and 1.6 times higher area under the concentration–time curve from 0–24 h (AUC0–24 h) in rats (oral dose of 300 mg/kg) for cilostazol and danazol, respectively. In conclusion, applying a wet media milling method with the combination of HPMC of a small molecular weight and Tween 80 as a dispersing agent, nanosuspensions can be practically prepared and conveniently utilized for enhancing the oral absorption of poorly water-soluble compounds in toxicology studies in the discovery stage.
The proportion of poorly water-soluble compounds among drug candidates in the discovery stage has been increasing for the past decade.1,2) Because of the poor solubility of these compounds, sufficient oral absorption is not observed in animal studies, and the evaluation of pharmacological and toxicological profiles of drug candidates has therefore become more challenging. In particular, this problem frequently occurs in toxicology studies that require high oral absorption and is now a common problem in pharmaceutical companies.3)
The application of formulation technologies such as solubilizing excipients,4) solid dispersions,5,6) and nanosuspensions7,8) is an attractive strategy for improving oral absorption of poorly water-soluble compounds. However, the use of solubilizing excipients and solid dispersions for toxicology studies in the discovery stage is practically limited because of toxicities of excipients and amount of drug substances for formulation studies, respectively. On the other hand, nanosuspensions have the potential to solve these problems. Because nanosuspensions can be prepared as highly drug-loaded formulations with small quantities of excipients, adverse effects caused by excipients can be avoided.9) In addition, nanosuspensions could reportedly be prepared with milligram-scale quantities of the drug substance using general wet media milling methods.10,11) Therefore, nanosuspensions are expected to be useful formulations for overcoming insufficient oral absorption in toxicology studies in the discovery stage.
However, further optimization of nanosuspension formulations is required so that these formulations can be conventionally used for discovery-stage toxicology studies. One requirement is that highly drug-concentrated nanosuspensions should be prepared with commonly used excipient types in proper quantities for toxicology studies. In addition, long-term stability of nanoparticles in suspension phases is required so that the nanosuspension formulations can be used for repeated dosing in toxicology studies. Although solidification techniques such as freeze-drying and spray-drying of nanosuspensions have been developed to avoid instability of nanoparticles in suspension phases,12,13) they are not favorable for toxicology studies because they normally require additional large quantities of excipients to redisperse the nanoparticles in water. In addition, nanosuspension formulations should be versatile for multiple compounds because many drug candidates are subjected to toxicology studies in the discovery stage.
Although various nanosuspension formulations have been reported,14,15) whether they can be prepared and can fulfil the abovementioned optimization requirements remains unclear. To the best of our knowledge, nanosuspension formulations that fulfil all these requirements have not been investigated previously. Several reports have shown that nanosuspensions could be prepared with high drug concentration and small quantities of excipients; however, they were prepared for only one compound.16–18) In contrast, other reports have shown that nanosuspensions of multiple compounds could be prepared with one formulation.11,19–21) However, these formulations are not suitable for our purpose because of one or more of the following reasons: they contain relatively large quantities of excipients; they contain unsuitable excipients for toxicology studies, such as ionic surfactants; or they have poor storage stability (or storage stability has not been investigated).
In the present study, we developed a practical method for preparing nanosuspension formulations for toxicology studies in the discovery stage with a focus on the following characteristics: i) containing a high drug concentration, ii) consisting of commonly used excipient types in proper quantities for toxicology studies, iii) having long-term stability, and iv) having versatility for use with diverse compounds. Excipients in dispersing agents are known to be important for reducing particle size and improving storage stability because they are adsorbed onto particle surfaces and inhibit crystal growth and aggregations in suspension phases.15,22) Therefore, effective dispersing agents to obtain the proposed nanosuspension formulation were screened using 11 different test compounds and wet media milling methods using a mixer mill (10 mg/batch) and a rotation/revolution mixer (0.5 g/batch). Furthermore, the crystalline properties and in vitro dissolution rates were evaluated for the nanosuspensions of two poorly water-soluble compounds, cilostazol and danazol, and their oral absorptions in rats at high doses were compared with those of microsuspensions. The practical applicability of nanosuspensions in toxicology studies in the discovery stage was assessed with respect to the method of formulation preparation and effect on oral absorption.
Experimental
MaterialsCilostazol, curucumin, furosemide, naproxen, phenytoin and nifedipine were purchased from Wako Pure Chemical Industries, Ltd. (Osaka, Japan). Danazol, spironolactone, cinnarizine, piroxicam and indomethacin were purchased from Sigma-Aldrich Co. (St. Louis, MO, U.S.A.). Three different grades of hydroxypropyl methylcellulose (HPMC) were purchased; TC-5E and Metolose 90SH-4000SR were purchased from Shin-Etsu Chemical Co., Ltd. (Tokyo, Japan) and the other grade of HPMC (Product number: H8384) was purchased from Sigma-Aldrich Co. These three HPMCs were classified by their viscosity in 2% aqueous solution at 20°C and referred to as HPMC 3 cP, 4000 cP, and 50 cP, respectively. Other polymers including methylcellulose (methylcellulose 400), which is referred to as MC (400 cP), and polyvinylpyrrolidone (Kollidone 17), which is referred to as PVP (K17), were purchased from Wako Pure Chemical Industries, Ltd. and BASF Japan Ltd. (Tokyo, Japan), respectively. Surfactants including Polysorbate 80 (Tween 80), sodium dodecyl sulfate (SDS), Cremophor RH40, Poloxamer 188, and D-α-tocopherol polyethylene glycol 1000 succinate (TPGS) were purchased from Sigma-Aldrich Co., Wako Pure Chemical Industries, Ltd., BASF Japan Ltd., MP biomedicals (Solon, OH, U.S.A.) and Eastman Chemical Co. (Kingsport, TN, U.S.A.), respectively. Sodium taurocholate and lecithin for a fasted state simulated intestinal fluid (FaSSIF, 3 mM sodium taurocholate and 0.75 mM lecithin in a pH 6.5 phosphate buffer) and a fed state simulated intestinal fluid (FeSSIF, 15 mM sodium taurocholate and 3.75 mM lecithin in a pH 5.0 phosphate buffer)23) were purchased from biorelevant.com (Croydon, England) and other reagents to prepare these solutions were purchased from Wako Pure Chemical Industries, Ltd.
Wet Media Milling Method (10 mg/batch)Ten milligrams of the test compound was weighted into a 2 mL Eppendorf Safe-Lock Tube (Eppendorf Co., U.S.A.) and milled at 30 Hz for 30 min by a mixer mill (MM301, Retsch GmbH, Germany) with 0.1 mL of the dispersing agent and 500 mg of zirconia beads with diameters of 0.3 mm (YTZ-0.3, Nikkato Co., Ltd., Japan). After milling, the tube was gently centrifuged at 3000 rpm (500×g) for 1 min to collect much of the content in the tube. Before the particle analyses, additional 0.9 mL of the dispersing agent was added and the tube was inversed several times to suspend all drug particles homogeneously. Then, being careful not to pipet zirconia beads, only the suspension phase was pipetting out for the subsequent particle analyses.
Wet Media Milling Method (0.5 g/batch)A half-gram of the test compound was weighed into a zirconia vessel and milled at 1700 rpm for 10 min with a chamber temperature of −20°C by a rotation/revolution mixer (NP-100, Thinky Corp., Japan) with 2.5 mL of the dispersing agent and 12.5 g of zirconia beads with diameters of 0.1 mm (YTZ-0.1, Nikkato Co., Ltd.). After that, 2.5 mL of the dispersing agent was added and milled at 400 rpm for 1–3 min (1 min for cilostazol and 3 min for the other test compounds) to deaggregate particle aggregations. Then the suspension was transferred into a nylon mesh (80 µm opening) chamber and gently centrifuged at 2000 rpm for 1 min to isolate the suspension from the zirconia beads. The suspension was poured into a polyethylene tube and the tube was inversed several times before the subsequent particle analyses.
Characterization of Particle SizeThe particle size distributions of the wet milled suspensions and the raw materials were analyzed by a laser diffractometer (Mastersizer 2000, Malvern Instruments, U.K.). The raw materials were dispersed in 0.5% HPMC (3 cP) before analyses. The measurement chamber was filled with water and adequate volume of the suspension was added. The Mie theory (dispersant refractive index=1.33; particle refractive index=1.62) was used for particle size calculation. The diameters at 50% and 90% of the cumulative volume distributions (D50 and D90, respectively) were determined. The particle size distributions of raw materials were analyzed after sonication for 30 s in the measurement chamber to estimate original particle size. The particle size stability of some wet milled suspensions at storage temperature of 5°C was characterized by measuring the particle size distributions over 4 weeks. The measurement was carried out in triplicate and the average and standard deviation were calculated.
In addition, the appearances of the milled particles were observed by a light microscope (VHX-500, KEYENCE, Japan) equipped with a VH-Z500 lens (KEYENCE). A drop of the suspension was put on a slide glass and pictures of the suspension were taken with 500× magnification.
Characterization of Crystalline PropertiesThe wet milled suspensions were frozen at −20°C and freeze dried with a trap temperature of −80°C using EYELA freeze dryer FDU-2100 (Tokyo Rikakikai Co., Ltd., Japan). Powder X-ray diffraction (PXRD) and differential scanning calorimetry (DSC) were measured for the freeze-dried nanoparticles and the raw materials. Both analyses were conducted in triplicate for each sample.
PXRD patterns were measured by X’ Pert Pro (Panalytical, the Netherlands). About 1 mg of each sample was placed onto a sample stage and PXRD patterns were collected by transmission mode with CuKα radiation at a voltage of 45 kV and a current of 40 mA. The samples were scanned over 2θ range at 3–40° with the scanning rate of 40°/min. The sample stage was oscillated during measurements to expose whole of the placed sample to X-ray. To reduce preferred orientation, PXRD patterns were collected 5 times with incident beam angle at −2°, −1°, 0°, 1°, and 2°, and then combined into one diffraction pattern.
DSC profiles were measured by X-DSC 7000 (SII Nanotechnology Inc., Japan). About 2 mg of each sample was placed in an aluminum pan and the pan was crimped before analysis. The heating program was carried out from 25°C to the end of melting at the heating rate of 10°C/min under nitrogen gas flow.
Saturation Solubility MeasurementSaturation solubility of each test compound at 37°C was determined in water, FaSSIF and FeSSIF. Excess amount of the raw material was added in each medium in glass vials. The vials were shaken continuously in a constant temperature chamber (37°C) for 24 h. Then 200 µL of each test medium was pipetted and filtered using a centrifugal filter unit (Ultrafree-MC with 0.45 µm polytetrafluoroethylene (PTFE) membrane filter, Millipore, U.S.A.) by centrifuging at 10000 rpm (5600×g) for 1 min. Each filtrate was pipetted and mixed to same volume of methanol. The concentration of the mixed solution was quantified by HPLC described as follows.
Quantitative analysis was performed by HPLC system (LC-20VP, Shimadzu Co., Kyoto, Japan) with the reverse-phase column (Capcell pak MG-II, 3.0 mm i.d.×35 mm, 3 µm, Shiseido Co., Ltd., Japan) at the column temperature of 40°C and the injection volume of 10 µL. Water containing 0.05% trifluoroacetic acid (A) and acetonitrile containing 0.05% trifluoroacetic acid (B) were used as the mobile phase, and the gradient condition was as follows: 5% B at 0 min, linear gradient to 100% B at 3.0 min, back to 5% B at 3.1 min and hold until 6.0 min with the total flow rate of 1.0 mL/min. The UV absorbance of each sample was detected at 257 nm and 286 nm for cilostazol and danazol, respectively.
Saturation solubility at room temperature was also measured for all of the test compounds by the same way as the above. The UV absorbance of each sample was detected at their maximum absorption wavelengths except for phenytoin and indomethacin. The UV absorbance of these compounds was detected at 220 nm.
In Vitro Dissolution StudyThe dissolution curves of the nanosuspensions and microsuspensions were estimated in 100 mL of FeSSIF at 37°C using a Distek Model 2100C Dissolution Test System (Distek, U.S.A.) with 100 mL glass vessels. The rotation speed of puddle was set to 50 rpm in this equipment. The nanosuspensions were prepared with 0.5% HPMC (3 cP)–0.5% Tween 80 by wet media milling and diluted to 10 mg/mL. The microsuspensions containing same drug concentration were prepared by a mortar and a pestle with 0.5% HPMC (3 cP)–0.5% Tween 80. The suspensions of 110 µL for cilstazol and 250 µL for danazol were added to the test mediums, and then aliquots of 200 µL were taken after 1, 5, 10, 20, 30, 40, 50, and 60 min. Each aliquot was filtered using a centrifugal filter unit (Ultrafree-MC with 0.1 µm polyvinylidene difluoride (PVDF) membrane filter, Millipore) by centrifuging at 10000 rpm (5600×g) for 1 min. According to a preliminary experiment and the previous report,24) a filter with a pore size of 0.1 µm was used to separate submicron sized particles from the solution phase. Each filtrate was pipetted and mixed to same volume of methanol. The concentration of the mixed solution was quantified by the HPLC method described above. The dissolution curves were measured in triplicate for each suspension and the average and standard deviation were calculated.
Toxicokinetic Study in Rats: FormulationFor microsuspensions, the test compound was ground with a small amount of the dispersing agent, 0.5% HPMC (3 cP)–0.5% Tween 80, using a mortar and a pestle, and mixed repeatedly to form a smooth paste. The paste was moved to a graduated cylinder and filled up to the required volume with the dispersing agent.
For nanosuspensions, 1 g of the test compound was weighed into a zirconia vessel and milled (at 1700 rpm for 10 min at −20°C) with 5 mL of the dispersing agent, 0.5% HPMC (3 cP)–0.5% Tween 80, and 20 g of zirconium beads with diameter of 0.1 mm using a rotation/revolution mixer. After that, 5 mL of the dispersing agent was added and particle aggregations were deaggregated by milling at 400 rpm for 1 min at −20°C. Then, the suspension was transferred to a nylon mesh (80 µm opening) chamber and centrifuged at 2000 rpm for 1 min to isolate the suspension from the zirconium beads. The obtained nanosuspension was diluted with the dispersing agent to make 60 mg/mL formulations.
Animal HandlingMale Crl:CD (Sprague-Dawley (SD)) rats (aged 6 weeks at dosing, body weight 170–192 g for cilostazol and 150–169 g for danazol) were supplied by Charles River Japan Inc. (Atsugi, Japan). The rats were acclimatized over a period of 5 d prior to dose administration. During the experiment period, rats were housed in group in plastic rat cages. Holding and study areas had automatic control of light cycles (07:00–19:00), temperature (20–26°C) and relative humidity (30–70%). A standard laboratory diet of known formulation (CRF-1, Oriental Yeast Co., Ltd., Japan) and domestic mains tap water were available ad libitum. The diet was not removed from the animals before administration. After the last sampling time of the blood, the rats were killed with carbon dioxide under anesthesia. All procedures in animal experiments were performed in accordance with the ‘Rules for Feeding and Storage of Experimental Animals and Animal Experiments’ of Mitsubishi Tanabe Pharma Corporation and under the Guidelines, based on the The Code of Ethics of the World Medical Association, issued (June 1, 2006) by the Ministry of Health, Labour and Welfare of Japan and Science Council of Japan.
Administration and Blood SamplingThree male rats were dosed orally as single dose by gastric gavage. The administered dose was 300 mg/kg. The dose volume was 5 mL/kg body weight.
Blood samples (0.2 mL) were collected from a subclavian vein in conscious animals and put into Ring Lock Microtubes (BM Equipment Co., Japan) containing air-dried heparin-Na. The samples were collected at 1, 2, 4, 7, and 24 h post-dose administration and immediately placed on ice and centrifuged at 12000 rpm (13200×g) for 2 min at 4°C to obtain plasma. The plasma was transferred to Eppendorf Safe-Lock Tubes. The plasma samples were stored frozen at −80°C until bioanalysis.
Bioanalytical MethodsQuantitative analysis of the total plasma concentration of cilostazol and danazol was performed by LC/MS-MS method using LC-10VP (Shimadzu Co.) and API3000 (AB SCIEX, U.S.A.) or LC-20VP (Shimadzu Co.) and API4000 (AB SCIEX). The internal standard of propranolol was added to each plasma sample, and then they were subjected to protein precipitated by addition of acetonitrile and centrifuged at 12000 rpm (13200×g) for 3–5 min at 4°C. Obtained supernatant was diluted with 0.1% formic acid aqueous solution and injected to LC/MS-MS at the injection volume of 10 µL. The mobile phases were 0.1% formic acid aqueous solution and acetonitrile (60 : 40, v/v) for cilostazol, 0.1% formic acid aqueous solution and methanol (40 : 60, v/v) for danazol. Separation was performed on Unison UK-C18 (2.0 mm i.d.×30 mm, 3 µm, Imtakt, U.S.A.) at the flow rate of 0.2 mL/min for cilostazol, Allure Basix (2.1 mm i.d.×100 mm, 5 µm, RESTEK, U.S.A.) at the flow rate of 0.4 mL/min for danazol. Detection was performed with positive electrospray ionization mode by monitoring the shift from precursor ions at m/z 370 to product ions of m/z 288 for cilostazol and precursor ions at m/z 338 to product ions of m/z 148 for danazol. Each lower limit of quantification in plasma was 0.01 µg/mL.
Toxicokinetic EvaluationIndividual plasma concentrations were calculated using Analyst version 1.4.2 (AB SCIEX). The Cmax, Tmax, and area under the concentration–time curve from 0 to 24 h (AUC0–24 h) values were determined in each animal using Phoenix WinNonlin Ver. 6.1 (Pharsight Co., U.S.A.) or Microsoft Excel 2003. The AUC0–24 h values were calculated according to the linear trapezoidal method. Below limit of quantitation was regarded as 0.000 µg/mL for the calculation of toxicokinetic parameters. The average and standard deviation were calculated and the toxicokinetic results are expressed as an average of three individuals (±S.D.).
Results
Screening Study of Nanosuspension FormulationsCellulose polymer derivatives and Tween 80 are commonly used excipients for preparing oral suspensions in toxicology studies25,26) and have recently been used for preparing nanosuspensions.14,15) In general, oral suspensions contain approximately 0.5% (w/v or w/w) of these excipients, and the dosing volumes of the suspensions range from 5 to 10 mL/kg in toxicology studies.27,28) Therefore, the following formulation conditions were used in the present study: cellulose polymer derivatives and Tween 80 were used as excipients for the dispersing agents; 0.5% (w/w) quantities of each excipient were used for the dispersing agents; and 100 mg/mL drug concentrations were used in the suspensions with the intention to deliver drug substances up to 1000 mg/kg. HPMC and MC were used as cellulose polymer derivatives, and three grades of HPMCs (3 cP, 50 cP, and 4000 cP) were used to vary the viscosities and molecular weights. In addition, Cremophor RH40, which is a commonly used excipient in toxicology studies,29) and PVP, Poloxamer 188, TPGS, and SDS, which are frequently used excipients for preparing nanosuspensions,14,15) were used in the present study to compare the effectiveness of nanosizing. However, it should be noted that normally, some of these excipients may not be used in toxicology studies. For example, SDS is known to raise gastrointestinal irritation.30,31)
A small-scale wet media milling method was used to screen effective dispersing agents for preparing the nanosuspensions. Cilostazol, which is a poorly water-soluble compound, was used as a test compound and milled in a mixer mill with various dispersing agents containing the abovementioned excipients. The effects of the dispersing agents were efficiently evaluated using a mixer mill because multiple batches (up to 20 batches) of the milligram-scale (10 mg/batch) drug substance could be simultaneously milled down to submicron size within 30 min. The particle sizes of the cilostazol suspensions prepared using this method are shown in Fig. 1. The D90 values of the particle size distributions were significantly large for all the suspensions prepared with only the polymers. The light microscope pictures shown in Fig. 2a also represented the presence of large particles in the suspension. In contrast, the surfactants appeared to be effective for reducing the particle size (Figs. 2b, c). The D90 values of the particle size distributions were decreased to <1 µm using 0.5% SDS and to approximately 1 µm using 0.5% Tween 80 and 0.5% TPGS. The many dispersing agents containing the polymers and the surfactants were also effective for reducing particle sizes. In the combination of the polymers and Tween 80, HPMC tended to produce smaller particles than other polymers. Because HPMC is known to cover a wide surface area of drug particles because of its open chain-like structure,32) milled particles might be effectively stabilized by HPMC. In the combination of HPMC and Tween 80, the smallest particle size was observed for HPMC (3 cP), which has the lowest viscosity and molecular weight in the three tested grades of HPMCs. From the results of the screening study using a mixer mill, it became apparent that the surfactants Tween 80, TPGS, and SDS were effective for preparing nanosuspensions and that smaller particles were obtained by combining them with HPMC (3 cP). Among these surfactants, Tween 80 seems to be the most commonly used excipient in toxicology studies because of its mild toxicity properties.25,27) We selected Tween 80 as well as HPMC for further formulation study.
Following this, some selected formulations were scaled up to 0.5 g/batch using a rotation/revolution mixer33) that was able to mill drug substances rapidly (approximately 2–15 min) at proper scale for toxicology studies in the discovery stage (approximately 0.1–10 g/batch). The dispersing agents of 0.5% Tween 80 and 0.5% HPMC (3 cP)–0.5% Tween 80 were used in the scale-up study. In addition, 0.5% HPMC (50 cP)–0.5% Tween 80 and 0.5% HPMC (4000 cP)–0.5% Tween 80 were selected to clearly compare the differences in the HPMC grades. The particle sizes of the milled suspensions are shown in Fig. 3a. The nanosuspensions could also be prepared with the selected dispersing agents at 0.5 g/batch, except for 0.5% HPMC (4000 cP)–0.5% Tween 80. The influences of viscosity and molecular weight may become more predominant in an intensive and rapid milling process of a rotation/revolution mixer. The smallest particle size was obtained for 0.5% HPMC (3 cP)–0.5% Tween 80, which was similar to the finding for the 10 mg/batch nanosuspensions.
The changes in the particle sizes of these nanosuspensions over 4 weeks at a storage temperature of 5°C were analyzed. The changes in the particle sizes are shown in Fig. 3b. The D90 values of the particle size distributions increased to micro size during the storage period for the nanosuspension prepared with 0.5% Tween 80 and 0.5% HPMC (50 cP)–0.5% Tween 80. On the other hand, the D90 value of the particle size distribution was preserved in submicron size for the nanosuspension prepared with 0.5% HPMC (3 cP)–0.5% Tween 80. It should be noted that the molecular weight of HPMC influenced not only particle size but also stability of nanosuspensions and only 0.5% HPMC (3 cP)–0.5% Tween 80 could stabilize cilostazol nanosuspensions long enough for toxicology studies.
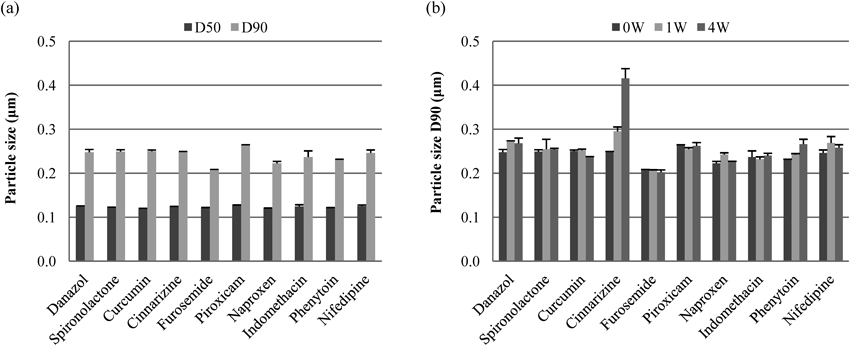
Application of Wet Media Milling with 0.5% HPMC (3 cP)–0.5% Tween 80 to Various CompoundsThe versatility of 0.5% HPMC (3 cP)–0.5% Tween 80 for effective use with various poorly water-soluble compounds was evaluated because a requirement for use of the proposed nanosuspension formulations in the discovery stage is that they can be simply prepared regardless of the drug substance. In addition to cilostazol, 10 different poorly water-soluble drug substances having a wide range of pKa values (neutral, acidic, and basic) and log P values were chosen as test compounds. The physicochemical properties of these compounds are listed in Table 1, and the particle sizes of the suspensions milled with 0.5% HPMC (3 cP)–0.5% Tween 80 in a rotation/revolution mixer are shown in Fig. 4a. Consequently, 100 mg/mL nanosuspensions could be prepared for all the test compounds. In addition, only small changes in the particle size distributions were observed for nine compounds over a 4-week period at 5°C (Fig. 4b). The particle size of the cinnarizine nanosuspension increased more than that of the other 9 compounds but remained in submicron size. Although it has been reported that the affinity of nonionic surfactants such as Tween 80, was weak for compounds having low log P values,21) log P values did not appear to have an influence on particle sizes in the present study.
Table 1. Physicochemical Properties of Test Compounds
| Compound | MW | Solubilitya) (µg/mL) | Melting point (°C) | c pKab) | c log P c) | Original particle size |
|---|
| D50 (µm) | D90 (µm) |
|---|
| Cilostazol | 369.46 | 3 | 159 | — | 3.5 | 18.1±0.3 | 55.1±0.2 |
| Danazol | 337.46 | <1 | 227 | — | 3.9 | 21.1±0.7 | 44.3±1.2 |
| Spironolactone | 416.57 | 28 | 206 | — | 2.8 | 4.8±0.3 | 9.1±0.7 |
| Curcumin | 368.38 | 2 | 182 | 7.9 (acid) | 2.9 | 20.0±0.6 | 38.1±1.3 |
| Cinnarizine | 368.51 | <1 | 120 | 8.4 (base) | 6.1 | 5.8±0.2 | 13.2±0.7 |
| Furosemide | 330.74 | 29 | 217 | 4.3 (acid) | 1.9 | 8.0±0.1 | 16.5±0.4 |
| Piroxicam | 331.35 | 14 | 201 | 1.8 (base), 4.3 (acid) | 1.9 | 16.4±0.6 | 29.2±1.9 |
| Naproxen | 230.26 | 29 | 156 | 4.2 (acid) | 2.8 | 14.1±0.4 | 31.3±0.9 |
| Indomethacin | 357.79 | 5 | 160 | 3.8 (acid) | 4.2 | 25.1±0.5 | 50.1±1.4 |
| Phenytoin | 252.27 | 20 | 297 | — | 2.3 | 12.3±0.1 | 21.8±0.2 |
| Nifedipine | 346.33 | 4 | 173 | — | 3.3 | 21.0±0.8 | 55.9±1.7 |
a) At room temperature. b) Calculated by ChemAxon Version 5.0. The strongest acid and base values are presented. c) Calculated by Daylight Version 4.9.
However, it should be noted that the particle size of cinnarizine was initially reduced to approximately 6 µm for D50 and 13 µm for D90 values of the particle size distribution before nanosuspension preparation. The raw material of cinnarizine had the largest particle size in the test compounds, which was approximately 50 µm for D50 and 90 µm for D90 values of particle size distribution. Because of the large particle size, the raw material of cinnarizine was not homogeneously nanosized by wet media milling with small-diameter zirconia beads (0.1 mm) over short times (10 min), and microsized particles were partly contaminated in the produced suspension (data not shown). Another problem occurred with piroxicum; the crystal form of the drug changed to the monohydrate form during wet media milling, which may have contributed to crystal growth. Therefore, the sample that was preliminarily changed to the monohydrate form by stirring in water/methanol was used for nanosuspension preparation.
Although the original particle size and the change in the crystal form should have been optimized before milling, stable nanosuspensions could be prepared with 0.5% HPMC (3 cP)–0.5% Tween 80 for the various compounds having various physicochemical properties. Consequently, it is thought that the proposed nanosuspension formulation can be simply prepared by general wet media milling methods with 0.5% HPMC (3 cP)–0.5% Tween 80 for a wide variety of drug substances.
Crystalline Properties of NanoparticlesCilostazol and danazol possess low solubility and high permeability properties (Table 2) and are categorized as BCS class II compounds. In addition, since they are neutral compounds, influence of pH can be ignored and effect of particle size on oral absorption will be more obvious. For these reasons, we selected them as model compounds for the toxicokinetic study in rats, as described below. To clearly estimate the oral absorption associated with each particle size, the differences in the crystal forms and crystallinities between the freeze-dried nanoparticles and the raw materials were analyzed by PXRD and DSC. The PXRD patterns and DSC profiles are shown in Figs. 5 and 6, respectively. From the PXRD patterns, changes in crystal forms after milling were not observed for either compound. In addition, from DSC profiles, changes in crystallinities were small for both compounds, as determined from the decreases in the heat of fusion of only approximately 5% (from 44.0±1.6 to 42.3±0.8 kJ/mol) and 10% (from 31.1±0.1 to 27.0±0.5 kJ/mol) for cilostazol and danazol, respectively. Although the melting points decreased by approximately 5°C (from 158.9±0.1 to 154.3±0.0°C) for cilostazol and 10°C (from 226.9±0.1 to 215.6±0.2°C) for danazol, they were thought to be mainly influenced by the reduction of particle sizes36) and coexistence of excipients. Consequently, it was thought that the crystalline properties of the nanoparticles of cilostazol and danazol would not influence oral absorption significantly.
Table 2. Saturation Solubility (Mean±S.D.,
n=3) and Apparent Permeability Coefficient (
Papp) of Caco-2 Monolayers
| Compound | Saturation solubility at 37°C (µg/mL) | Papp (×10−5 cm/s) |
|---|
| Cilostazol | Water | 4.5±0.1 | 1.92±—a) |
| FaSSIF | 7.5±0.0 | |
| FeSSIF | 16.5±0.1 | |
| Danazol | Water | 0.1±0.0 | 8.73±4.98b) |
| FaSSIF | 6.0±0.1 | |
| FeSSIF | 44.7±4.9 | |
a) Toyobuku et al.34) b) Takano et al.35)
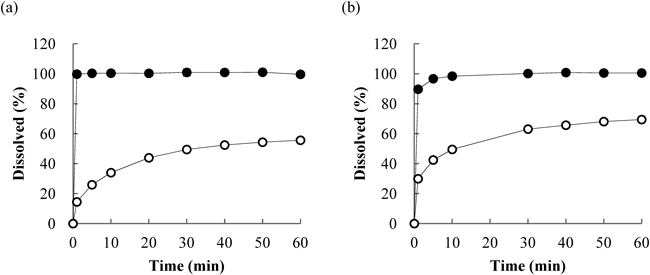
An in vitro dissolution study was conducted for the nanosuspensions and microsuspensions of cilostazol and danazol. The D50 values of the particle size distributions of these nanosuspensions and microsuspensions were 0.13 µm and 16 µm for cilostazol and 0.15 µm and 7 µm for danazol, respectively. The toxicokinetic study in rats was conducted in the nonfasted state, and Hagio et al.37) reported that the concentration of bile micelles was approximately 9–17 mM in the jejunum and 15–25 mM in the ileum in nonfasted rats. Hence, FeSSIF, which contains 15 mM of bile micelles (sodium taurocholate), was used as a test medium in the in vitro dissolution study.
The dissolution curves are shown in Fig. 7. The dissolutions from the microsuspensions of both compounds were slow, and added samples were not dissolved during 1 h. On the other hand, the dissolution rates were dramatically improved by the nanosuspensions for both compounds, and all the added samples were dissolved within 1–5 min.
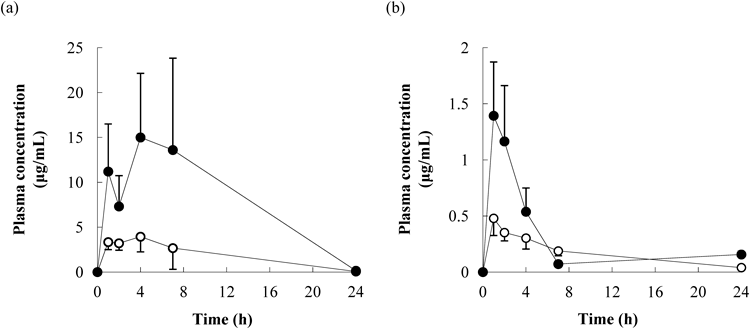
Effect on Oral Absorption of High-Dose NanosuspensionsThe nanosuspensions and microsuspensions of cilostazol and danazol were orally administered to rats at 300 mg/kg (60 mg/mL at 5 mL/kg). The D50 values of the particle size distributions of the nanosuspensions and microsuspensions were 0.13 µm and 17 µm for cilostazol and 0.13 µm and 7 µm for danazol, respectively. The plots of the mean plasma concentrations versus time are shown in Fig. 8. The toxicokinetic parameters are presented in Table 3.
Table 3. Toxicokinetic Parameters Following Oral Administration of Cilostazol and Danazol to Rats
| Dose (mg/kg) | Formulation | Tmax (h) | Cmax (µg/mL) | AUC0–24 h (µg·h/mL) |
|---|
| Cilostazol |
| 300 | Microsuspension | 3.3±1.2 | 4.1±1.6 | 45.2±29.6 |
| 300 | Nanosuspension | 5.0±1.7 | 15.9±8.5 | 197.0±127.0 |
| Danazol |
| 300 | Microsuspension | 1.0±0.0 | 0.5±0.2 | 4.0±0.8 |
| 300 | Nanosuspension | 1.3±0.6 | 1.5±0.5 | 6.5±1.4 |
Systemic exposure (Cmax or AUC0–24 h) in rat plasma was higher for the nanosuspensions than for the microsuspensions for both the compounds. For the plasma concentrations of cilostazol, Cmax and AUC0–24 h were 3.9 and 4.4 times higher for the nanosuspension, respectively. For danazol, Cmax and AUC0–24 h were 3.0 and 1.6 times higher for the nanosuspension, respectively. Although AUC0–24 h for danazol was not significantly enhanced by the nanosuspensions, higher Cmax values were observed, which was important in the evaluation of Cmax-related toxicity. From the results of the in vivo study, the nanosuspensions were considered to be effective for improving oral absorption even at high doses.
Discussion
The proportion of poorly water-soluble compounds among drug candidates in the discovery stage has been increasing, and insufficient oral absorption has been frequently observed for these compounds, particularly in toxicology studies. Using formulation technologies to address this problem, we developed a practical method for preparing nanosuspension formulations for toxicology studies in the discovery stage and assessed their applicability and effects on oral absorption at high doses.
From the results of the formulation study, we found that nanosuspensions with high drug concentrations (100 mg/mL) could be simply prepared with 0.5% HPMC (3 cP)–0.5% Tween 80, which consists of commonly used excipient types in proper quantities for toxicology studies. Although small quantities of excipients were used with high concentrations of drug substances, stable nanosuspensions could be prepared for all the 11 test compounds. Therefore, the applicable range of nanosuspension formulations can be expanded using the dispersing agent that we discovered in the present study. Nanosuspensions can be simply and rapidly prepared on small scales using general wet media milling methods with 0.5% HPMC (3 cP)–0.5% Tween 80 and then conventionally applied to toxicology studies from the discovery stage.
Since both HPMC (3 cP) and Tween 80 are amphiphilic compounds, they could adsorb onto surface of the milled particles and stabilize them in the aqueous suspension phases. In addition, their molecular weights might be adequate for preparing nanoparticles and improving storage stability. Tween 80 was beneficial for reducing particle size during the milling process, while large particles were observed when only HPMC (3 cP) was used as a dispersing agent. The rate of adsorption of small molecular excipients onto drug particles has been shown to be faster than that of large molecular excipients.19,38) Hence, it was thought that Tween 80, which has a molecular weight approximately one-tenth that of HPMC (3 cP), contributed to stabilizing particles during the milling process by its rapid adsorption onto the drug particles, which resulted in the formation of nanosuspensions. On the other hand, HPMC (3 cP) was effective for stabilizing nanoparticles during storage. Although the rate of adsorption of large molecular excipients was slow, their adsorption was known to be more difficult to reverse.39) Hence, HPMC (3 cP) was thought to contribute to the long-term stability of nanoparticles because of its adsorption characteristic.
The combination of HPMC and Tween 80 was previously applied as a dispersing agent for preparing nanosuspensions.11,40) However, the molecular weight of HPMC used in those studies was larger than HPMC (3 cP), the viscosity grades were from 50 to 4000 cP. In the present study, we found that HPMC (3 cP), which has the lowest viscosity and the smallest molecular weight of the tested HPMCs, produced smaller particle size and more stable nanosuspensions than any other HPMCs (such as 50 cP and 4000 cP) by wet media milling with Tween 80. When the molecular weight of excipients is too large, the amount of adsorbed excipients onto drug particles will be lower because of their slow adsorption rate. The molecular weight of HPMC (3 cP) is approximately 16000, and excipients having molecular weights of 5000–25000 have been reported to be long enough for steric repulsion and thus stabilization of nanoparticles.41) HPMC (3 cP) should possess suitable property about adsorption onto drug particles during milling and stabilizing them for a long time. Using common excipient types with adequate molecular weights was considered to be important to obtain stable nanosuspensions that were usable in toxicology studies. In the present study, the combination of HPMC (3 cP) and Tween 80 was found to be the most suitable dispersing agent for the intended purpose.
In terms of in vivo application of nanosuspensions, although oral absorption is considered to be limited by saturation solubility rather than by dissolution rate at high doses,42) our results revealed that nanosuspensions were effective even at 300 mg/kg for both cilostazol and danazol, in accordance with previous reports that nanosuspensions improved oral absorption in high-dose in vivo studies.43–46)
The degree of enhancement of AUC0–24 h by the use of nanosuspensions was higher for cilostazol than for danazol, although the in vitro dissolution rates were increased similarly, and the changes in crystalline properties were not significant for either compound. Sugano47) assumed that intestinal membrane permeability could be improved by nanosuspensions because nanoparticles permeate through the unstirred water layer, which is adjacent to the intestinal membrane. Moreover, bile micelles that include drug molecules in the intestinal tract are known to permeate through the unstirred water layer.48,49) Therefore, intestinal membrane permeability can be improved by both nanoparticles and bile micelles. In addition, the concentration of bile micelles is known to be relatively high in the intestinal tract of rats, and it was found to be approximately 9–17 mM in the jejunum and 15–25 mM in the ileum in the nonfasted state37) and approximately 50 mM in the upper jejunum and 100 mM in the lower jejunum in the fasted state.50) Hence, the effect of nanosuspensions on oral absorption in rats may become ambiguous for compounds such as danazol that have high solubility in bile micelles. In contrast, the probability of improving the rate-limiting step in oral absorption using nanosuspensions for compounds such as cilostazol that have low solubility in bile micelles should be high. The relationship between the effect of nanoparticles on oral absorption at high doses and physicochemical characters, including solubility in bile micelles, should be further investigated in the future.
Based on these results, nanosuspensions can be used in toxicology studies in the discovery stage because of the simple method of formulation preparation that uses common excipients and the improvement of oral absorption at high doses. Nanosuspensions can be a convenient formulation for enhancing in vivo exposures of poorly water-soluble compounds in the discovery stage.
Conclusion
Although there continues to be a great need to enhance oral absorption of poorly water-soluble compounds in toxicology studies in the discovery stage, the quantities of drug substances required for formulation studies and the adverse effects caused by excipients have been considered to be bottlenecks for applying formulation technologies. In the present study, we optimized formulations of nanosuspensions and found that a suitable nanosuspension formulation for the intended purpose could be simply prepared by a wet media milling method with the combination of HPMC of a small molecular weight and Tween 80 as a dispersing agent. Furthermore, the nanosuspensions enabled superior oral absorption in rats even at high doses. This nanosuspension preparation method does not lead to effort for toxicological evaluations in the discovery stage and can reduce the time required for candidate selection.
Acknowledgment
The authors are grateful to Masayuki Ube and Hiroshi Sakurada (Safety Research Laboratories, Mitsubishi Tanabe Pharma Corporation) for the toxicokinetic study, Michihiro Kono and Koji Takekawa (Safety Research Laboratories, Mitsubishi Tanabe Pharma Corporation) for bioanalysis and toxicokinetic evaluation, and Mimie Takano and Harumi Hayashi (Safety Research Laboratories, Mitsubishi Tanabe Pharma Corporation) for technical assistance.
References
- 1) Lipinski C. A., J. Pharmacol. Toxicol. Methods, 44, 235–249 (2000).
- 2) Lipinski C. A., Am. Pharm. Rev., 5, 82–85 (2002).
- 3) Higgins J., Cartwright M. E., Templeton A. C., Drug Discov. Today, 17, 828–836 (2012).
- 4) Strickley R. G., Pharm. Res., 21, 201–230 (2004).
- 5) Vasconcelos T., Sarmento B., Costa P., Drug Discov. Today, 12, 1068–1075 (2007).
- 6) Van den Mooter G., Drug Discov. Today. Technol., 9, e79–e85 (2012).
- 7) Junghanns J. U., Müller R. H., Int. J. Nanomedicine, 3, 295–309 (2008).
- 8) Merisko-Liversidge E., Liversidge G. G., Adv. Drug Deliv. Rev., 63, 427–440 (2011).
- 9) Möschwitzer J. P., Int. J. Pharm., 453, 142–156 (2013).
- 10) Van Eerdenbrugh B., Stuyven B., Froyen L., Van Humbeeck J., Martens J. A., Augustijns P., Van den Mooter G., AAPS PharmSciTech, 10, 44–53 (2009).
- 11) Niwa T., Miura S., Danjo K., Int. J. Pharm., 405, 218–227 (2011).
- 12) Van Eerdenbrugh B., Froyen L., Martens J. A., Blaton N., Augustijns P., Brewster M., Van den Mooter G., Int. J. Pharm., 338, 198–206 (2007).
- 13) Niwa T., Miura S., Danjo K., Pharm. Res., 28, 2339–2349 (2011).
- 14) Van Eerdenbrugh B., Van den Mooter B., Augustijns P., Int. J. Pharm., 364, 64–75 (2008).
- 15) Wu L., Zhang J., Watanabe W., Adv. Drug Deliv. Rev., 63, 456–469 (2011).
- 16) Jacobs C., Kayser O., Müller R. H., Int. J. Pharm., 196, 161–164 (2000).
- 17) Ain-Ai A., Gupta P. K., Int. J. Pharm., 351, 282–288 (2008).
- 18) Pardeike J., Müller R. H., Int. J. Pharm., 391, 322–329 (2010).
- 19) Lee J., Choi J.-Y., Park C. H., Int. J. Pharm., 355, 328–336 (2008).
- 20) Van Eerdenbrugh B., Vermant J., Martens J. A., Froyen L., Van Humbeeck J., Augustijns P., Van Den Mooter G., J. Pharm. Sci., 98, 2091–2103 (2009).
- 21) George M., Ghosh I., Eur. J. Pharm. Sci., 48, 142–152 (2013).
- 22) Peltonen L., Hirvonen J., J. Pharm. Pharmacol., 62, 1569–1579 (2010).
- 23) Galia E., Nicolaides E., Horter D., Lobenberg R., Reppas C., Dressman J. B., Pharm. Res., 15, 698–705 (1998).
- 24) Juenemann D., Jantratid E., Wagner C., Reppas C., Vertzoni M., Dressman J. B., Eur. J. Pharm. Biopharm., 77, 257–264 (2011).
- 25) Neervannan S., Expert Opin. Drug Metab. Toxicol., 2, 715–731 (2006).
- 26) Li P., Zhao L., Int. J. Pharm., 341, 1–19 (2007).
- 27) Gad S. C., Cassidy C. D., Aubert N., Spainhour B., Robbe H., Int. J. Toxicol., 25, 494–521 (2006).
- 28) Enright B. P., McIntyre B. P., Thackaberry E. A., Treinen K. A., Kopytek S. J., Birth Defects Res. B. Dev. Reprod. Toxicol., 89, 504–516 (2010).
- 29) Pestel S., Martin H.-J., Maier G.-M., Guth B., J. Pharmacol. Toxicol. Methods, 54, 200–214 (2006).
- 30) Nissim J. A., Nature (London), 187, 305–307 (1960).
- 31) Nakamura J., Takada S., Ueda S., Hamaura T., Yamamoto A., Kimura T., Sezaki H., Chem. Pharm. Bull., 33, 3527–3529 (1985).
- 32) Verma S., Huey B. D., Burgess D. J., Langmuir, 25, 12481–12487 (2009).
- 33) Takatsuka T., Endo T., Jianguo Y., Yuminoki K., Hashimoto N., Chem. Pharm. Bull., 57, 1061–1067 (2009).
- 34) Toyobuku H., Tamai I., Ueno K., Tsuji A., J. Pharm. Sci., 92, 2249–2259 (2003).
- 35) Takano R., Sugano K., Higashida A., Hayashi Y., Machida M., Aso Y., Yamashita S., Pharm. Res., 23, 1144–1156 (2006).
- 36) Sun W., Mao S., Shi Y., Li L. C., Fang L., J. Pharm. Sci., 100, 3365–3373 (2011).
- 37) Hagio M., Matsumoto M., Fukushima M., Hara H., Ishizuka S., J. Lipid Res., 50, 173–180 (2009).
- 38) Duro R., Souto C., Gomez-Amoza J. L., Martinez-Pacheco R., Concheiro A., Drug Dev. Ind. Pharm., 25, 817–829 (1999).
- 39) Verma S., Kumar S., Gokhale R., Burgess D. J., Int. J. Pharm., 406, 145–152 (2011).
- 40) Kumar M. P., Rao Y. M., Apte S., Curr. Nanosci., 3, 191–194 (2007).
- 41) Lee J., Lee S.-J., Choi J.-Y., Yoo J. Y., Ahn C.-H., Eur. J. Pharm. Sci., 24, 441–449 (2005).
- 42) Butler J. M., Dressman J. B., J. Pharm. Sci., 99, 4940–4954 (2010).
- 43) Kesisoglou F., Panmai S., Wu Y., Adv. Drug Deliv. Rev., 59, 631–644 (2007).
- 44) Sigfridsson K., Nordmark A., Theilig S., Lindahl A., Drug Dev. Ind. Pharm., 37, 185–192 (2011).
- 45) Sigfridsson K., Lundqvist A. J., Strimfors M., Drug Dev. Ind. Pharm., 37, 243–251 (2011).
- 46) Kesisoglou F., Mitra A., AAPS J., 14, 677–687 (2012).
- 47) Sugano K., Int. J. Pharm., 387, 103–109 (2010).
- 48) Amidon G. E., Higuchi W. I., Ho N. F., J. Pharm. Sci., 71, 77–84 (1982).
- 49) Sugano K., Int. J. Pharm., 368, 116–122 (2009).
- 50) Tanaka Y., Hara T., Waki R., Nagata S., J. Pharm. Pharm. Sci., 15, 510–518 (2012).