Experimental
1H-NMR spectra were recorded on a Varian VNS-400, JEOL JNM-LA400, or JEOL JNM-AL400 and the chemical shifts were expressed in δ (ppm) values with tetramethylsilane as an internal reference (s=singlet, d=doublet, t=triplet, m=multiplet, dd=doublet of doublets, tt=triplet of triplets, and br=broad peak). Mass spectra (MS) were recorded on Thermo Electron LCQ Advantage. Elemental analyses were performed using Yanaco MT-6 (C, H, N), Elementar Vario EL III (C, H, X), and Dionex ICS-3000 (S, halogene) and were within ±0.4% of theoretical values. Electrospray ionization (ESI) positive high resolution mass spectrum (HR-MS) was obtained using Waters LCT Premier.
tert-Butyl 4-(Pyridin-4-ylacetyl)piperidine-1-carboxylate (5)Under argon gas atmosphere, to a mixture of 4-methylpyridine (2.63 g, 28.3 mmol) in tetrahydrofuran (THF) (50 mL) cooled with ice-water bath was added lithium diisopropylamide (2.0 M solution in THF/heptane/ethylbenzene; 17.0 mL, 34.0 mmol), and the mixture was stirred at same temperature for 30 min. To the resultant mixture cooled with dryice-acetone bath was slowly added a solution of tert-butyl 4-(N-methoxy-N-methylcarbamoyl)-1-piperidinecarboxylate (4, 5.12 g, 18.8 mmol) in THF (100 mL), and the mixture was stirred at same temperature for 1 h. The mixture was quenched with water and extracted with EtOAc. The organic layer was washed with brine, dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by silica gel column chromatography (10–50% EtOAc in CHCl3) to give 5 (4.05 g, 71%) as a yellow oil. 1H-NMR (CDCl3) δ: 1.45 (9H, s), 1.50–1.63 (2H, m), 1.82 (2H, br d, J=12.2 Hz), 2.58 (1H, tt, J=11.2, 3.9 Hz), 2.78 (2H, br t, J=11.2 Hz), 3.76 (2H, s), 4.03–4.21 (2H, m), 7.11–7.14 (2H, m), 8.55–8.58 (2H, m); MS (ESI) m/z 305 [M+H]+.
1-Methyl-4-(pyridin-4-yl)-1H-pyrazol-3-ol (13)To a solution of ethyl 4-pyridineacetate (12, 25.0 g, 151 mmol) in DMF (100 mL) was added DMF-DMA (50.4 mL, 378 mmol), and the mixture was stirred at 80°C for 2 h. The reaction was cooled at room temperature and concentrated in vacuo. To the residue in ethanol (250 mL) cooled with ice-water bath were added methylhydrazine (15.9 mL, 302 mmol) and acetic acid (60 mL), and the mixture was stirred at room temperature for 16 h. The mixture was concentrated in vacuo, and the residue was purified by silica gel column chromatography (0–10% MeOH in CHCl3) to give crude product, which was washed with EtOAc–hexane to give an orange solid. The orange solid was purified again by silica gel column chromatography (0–10% MeOH in CHCl3) to give 13 (15.3 g, 58%) as a pale yellow solid. 1H-NMR (DMSO-d6) δ: 3.67 (3H, s), 7.55–7.59 (2H, m), 8.09 (1H, s), 8.39–8.44 (2H, m), 10.71 (1H, br s); MS (ESI) m/z 176 [M+H]+.
tert-Butyl 4-[1-Methyl-4-(pyridin-4-yl)-1H-pyrazol-3-yl]piperidine-1-carboxylate (6)Compound 6 was prepared from 5 in a manner similar to that described for compound 13, with a yield of 20% as a colorless solid. 1H-NMR (CDCl3) δ: 1.46 (9H, s), 1.72–1.89 (4H, m), 2.66–2.90 (2H, m), 2.93–3.04 (1H, m), 3.90 (3H, s), 4.19 (2H, br s), 7.22–7.25 (2H, m), 7.47 (1H, s), 8.56–8.61 (2H, m); MS (ESI) m/z 343 [M+H]+.
4-[1-Methyl-3-(piperidin-4-yl)-1H-pyrazol-4-yl]pyridine Dihydrochloride (7)To a mixture of 6 (342 mg, 1.00 mmol) in MeOH (7.3 mL) was added 4 M HCl/dioxane (3.6 mL, 14.4 mmol), and the mixture was stirred at 50°C for 1 h. After cooling at room temperature, the mixture was concentrated in vacuo to give 7 (315 mg, quant) as a pale yellow solid. 1H-NMR (DMSO-d6) δ: 1.88–2.06 (4H, m), 3.00–3.12 (2H, m), 3.26–3.50 (3H, m), 3.90 (3H, s), 8.07 (2H, d, J=6.7 Hz), 8.58 (1H), 8.78 (2H, d, J=6.7 Hz), 8.97 (1H, br s), 9.24 (1H, br s); MS (ESI) m/z 398 [M+H]+.
2-(2-{4-[1-Methyl-4-(pyridin-4-yl)-1H-pyrazol-3-yl]piperidin-1-yl}ethyl)quinoline Trihydrochloride (8)To a solution of 7 (100 mg, 0.32 mmol) in MeOH (30 mL) was added triethylamine (90 µL, 0.65 mmol), and the mixture was concentrated in vacuo. To the residue in ethanol (2.0 mL) were added 2-vinylquinoline (100 mg, 0.65 mmol) and acetic acid (30 µL, 0.53 mmol), and the mixture was stirred at 80°C for 12 h. After cooling at room temperature, the mixture was partitioned between saturated NaHCO3 aqueous solution and EtOAc. The organic layer was washed with brine, dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by silica gel column chromatography (1–10% MeOH in CHCl3) to give a pale brown solid, which was dissolved in MeOH (5.0 mL) and 4 M HCl/dioxane (0.25 mL, 1.00 mmol) was added to the mixture. The mixture was concentrated in vacuo, and the residue was washed with diisopropylether to give 8 (21 mg, 13%) as a dark green solid. 1H-NMR (DMSO-d6) δ: 2.04–2.26 (4H, m), 3.27 (2H, br s), 3.45 (1H, br s), 3.69 (6H, br s), 3.91 (3H, s), 7.71–7.80 (2H, m), 7.90–7.97 (1H, m), 8.07 (2H, d, J=6.6 Hz), 8.13 (1H, d, J=8.0 Hz), 8.21 (1H, d, J=8.3 Hz), 8.58 (1H, s), 8.68 (1H, d, J=8.0 Hz), 8.79 (2H, d, J=6.6 Hz); MS (ESI) m/z 398 [M+H]+. Anal. Calcd for C25H27N5·3HCl·4.3H2O·0.3C6H14O: C, 52.34; H, 7.01; N, 11.39; Cl, 17.29. Found: C, 52.44; H, 6.81; N, 11.09; Cl, 16.98.
2-({4-[1-Methyl-4-(pyridin-4-yl)-1H-pyrazol-3-yl]piperidin-1-yl}methyl)quinoline Trihydrochloride (9)To a mixture of 7 (144 mg, 0.46 mmol) and 2-(chloromethyl)quinoline (100 mg, 0.47 mmol) in DMF (8.0 mL) was added N-ethyl-N-isopropylpropan-2-amine (0.33 mL, 1.90 mmol), and the mixture was stirred at 60°C for 4 h. After cooling at room temperature, the mixture was diluted with EtOAc and washed with saturated NaHCO3 aqueous solution and brine, dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by silica gel column chromatography (1–9% MeOH in CHCl3) to give a pale yellow solid, which was dissolved in MeOH (5 mL) and 4 M HCl/EtOAc (0.35 mL) was added to the mixture. The mixture was concentrated in vacuo to give 9 (47 mg, 21%) as a beige solid. 1H-NMR (DMSO-d6) δ: 2.04–2.29 (4H, m), 3.28–3.47 (3H, m), 3.53–3.63 (2H, m), 3.99 (3H, s), 4.67 (2H, s), 7.70 (1H, dd, J=7.2, 7.2 Hz), 7.82–7.89 (1H, m), 7.94 (1H, d, J=8.5 Hz), 8.02–8.13 (4H, m), 8.54 (1H, d, J=8.5 Hz), 8.58 (1H, s), 8.77 (2H, d, J=6.7 Hz); MS (ESI) m/z 384 [M+H]+. Anal. Calcd for C24H25N5·3HCl·4.1H2O: C, 50.86; H, 6.44; N, 12.36; Cl, 18.77. Found: C, 51.01; H, 6.41; N, 12.24; Cl, 18.56.
[4-(Quinolin-2-yl)phenyl]methanol (11)Under argon gas atmosphere, to a mixture of 2-chloroquinoline (1.05 g, 6.42 mmol), [4-(hydroxymethyl)phenyl]boronic acid (10, 976 mg, 6.42 mmol) and Pd(PPh3)4 (384 mg, 0.33 mmol) in 1,2-dimethoxyethane (15 mL) and water (5 mL) was added Na2CO3 (1.65 g, 15.6 mmol), and the mixture was stirred at 100°C for 14 h. After cooling at room temperature, the mixture was partitioned between EtOAc and water. The organic layer was dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by silica gel column chromatography (10–100% EtOAc in hexane) to give 11 (1.50 g, 99%) as an off-white solid. 1H-NMR (DMSO-d6) δ: 4.60 (2H, d, J=5.7 Hz), 5.28 (1H, t, J=5.7 Hz), 7.50 (2H, d, J=8.2 Hz), 7.56–7.62 (1H, m), 7.75–7.61 (1H, m), 7.99 (1H, d, J=8.1 Hz), 8.07 (1H, d, J=8.5 Hz), 8.14 (1H, d, J=8.7 Hz). 8.25 (2H, d, J=8.2 Hz), 8.44 (1H, d, J=8.7 Hz); MS (ESI) m/z 236 [M+H]+.
[3-(Quinolin-2-yl)phenyl]methanol (15e)Compound 15e was prepared from [3-(hydroxymethyl)phenyl]boronic acid (19) and 2-chloroquinoline in a manner similar to that described for compound 11, with a quantitative yield as a yellow oil. 1H-NMR (CDCl3) δ: 2.13 (1H, br s), 4.81 (2H, br s), 7.43–7.56 (3H, m), 7.71–7.76 (1H, m), 7.83 (1H, d, J=8.1 Hz), 7.87 (1H, d, J=8.6 Hz), 8.03–8.07 (1H, m), 8.16–8.24 (3H, m); MS (ESI) m/z 236 [M+H]+.
2-[3-({[1-Methyl-4-(pyridin-4-yl)-1H-pyrazol-3-yl]oxy}methyl)phenyl]quinoline Dihydrochloride (16e)To a mixture of 13 (200 mg, 1.14 mmol), 15e (295 mg, 1.26 mmol) and tributylphosphine (346 mg, 1.71 mmol) in THF (20 mL) was added 1,1′-(azodicarbonyl)dipiperidine (ADDP; 432 mg, 1.71 mmol), and the mixture was stirred at room temperature for 14 h before the mixture was concentrated in vacuo. The residue was suspended in EtOAc, and the insoluble material was removed by filtration. The filtrate was concentrated in vacuo, and the residue was purified by silica gel column chromatography (0–3% MeOH in CHCl3) to give a colorless oil, which was dissolved in EtOAc (20 mL). To the mixture was added 4 M HCl/EtOAc (1.1 mL) and the precipitate was collected by filtration give 16e (289 mg, 54%) as an off-white solid. 1H-NMR (DMSO-d6) δ: 3.85 (3H, s), 5.55 (2H, s), 7.63–7.75 (3H, m), 7.86–7.92 (1H, m), 8.11 (1H, s, J=8.1 Hz), 8.15 (2H, d, J=7.0 Hz), 8.23–8.31 (3H, m), 8.45 (1H, s), 8.67 (1H, d, J=8.7 Hz), 8.70–8.74 (3H, m); MS (ESI) m/z 393 [M+H]+. Anal. Calcd for C25H20N4O·2HCl·0.9H2O: C, 62.35; H, 4.98; N, 11.63; Cl, 14.72. Found: C, 62.70; H, 5.31; N, 11.45; Cl, 14.83.
2-[4-({[1-Methyl-4-(pyridin-4-yl)-1H-pyrazol-3-yl]oxy}methyl)phenyl]quinoline Dihydrochloride (14)Compound 14 was prepared from 11 and 13 in a manner similar to that described for compound 16e, with a yield of 39% as an off-white solid. 1H-NMR (DMSO-d6) δ: 3.84 (3H, s), 5.52 (2H, s), 7.66–7.72 (1H, m), 7.74 (2H, d, J=8.3 Hz), 7.85–7.91 (1H, m), 8.10 (1H, d, J=8.1 Hz), 8.15 (2H, d, J=7.0 Hz), 8.20–8.27 (2H, m), 8.34 (2H, d, J=8.3 Hz), 8.64 (1H, d, J=8.7 Hz), 8.71 (1H, s), 8.74 (2H, d, J=7.0 Hz) ; MS (ESI) m/z 393 [M+H]+. Anal. Calcd for C25H20N4O·2HCl·3.1H2O: C, 57.61; H, 5.45; N, 10.75; Cl, 13.60. Found: C, 57.76; H, 5.51; N, 10.76; Cl, 13.54.
[1-(Quinolin-2-yl)piperidin-4-yl]methanol (15d)To a mixture of 2-chloroquinoline (2.50 g, 15.3 mmol) and piperidine-4-ylmethanol (18, 5.28 g, 45.8 mmol) in DMF (20 mL) was added K2CO3 (3.17 g, 22.9 mmmol), and the mixture was stirred at 110°C for 1 d. After cooling at room temperature, the mixture was concentrated in vacuo. The residue was partitioned between EtOAc and water, and the organic layer was concentrated in vacuo. The residue was purified by silica gel column chromatography (20–50% EtOAc in hexane) to give 15e (3.69 g, quant) as a pale yellow solid. 1H-NMR (DMSO-d6) δ: 1.08–1.20 (2H, m), 1.62–1.80 (3H, m), 2.84–2.92 (2H, m), 3.28 (2H, t, J=5.8 Hz), 4.47 (1H, t, J=5.3 Hz), 4.51–4.58 (2H, m), 7.15–7.21 (1H, m), 7.23 (1H, d, J=9.2 Hz), 7.46–7.56 (2H, m), 7.64–7.68 (1H, m), 7.98 (1H, d, J=9.2 Hz); MS (ESI) m/z 243 [M+H]+.
3-[Methyl(Quinolin-2-yl)amino]propan-1-ol (15c)Compound 15c was prepared from 17 and 2-chloroquinoline in a manner similar to that described for compound 15d, with a yield of 68% as a colorless oil. 1H-NMR (DMSO-d6) δ: 1.70–1.78 (2H, m), 3.13 (3H, s), 3.42–3.48 (2H, m), 3.67 (2H, t, J=7.0 Hz), 4.69 (1H, t, J=5.3 Hz), 7.07 (1H, d, J=9.2 Hz), 7.13–7.18 (1H, m), 7.46–7.52 (2H, m), 7.64–7.68 (1H, m), 7.99 (1H, d, J=9.1 Hz); MS (ESI) m/z 217 [M+H]+.
2-(3-{[1-Methyl-4-(pyridin-4-yl)-1H-pyrazol-3-yl]oxy}propyl)quinoline Dihydrochloride (16a)Compound 16a was prepared from 13 and 15a in a manner similar to that described for compound 16e, with a yield of 41% as a yellow oil. 1H-NMR (DMSO-d6) δ: 2.40–2.50 (2H, m), 3.47–3.55 (2H, m), 3.79 (3H, s), 4.46 (2H, t, J=5.8 Hz), 7.82 (1H, t, J=7.6 Hz), 7.94–8.09 (4H, m), 8.19 (1H, d, J=8.3 Hz), 8.46 (1H, brd, J=8.3 Hz), 8.59 (1H, s), 8.64 (2H, d, J=7.0 Hz), 8.92 (1H, br d, J=8.3 Hz); MS (ESI) m/z 345 [M+H]+. Anal. Calcd for C21H20N4O·2HCl·3H2O: C, 53.51; H, 5.99; N, 11.89; Cl, 15.04. Found: C, 53.56; H, 6.03; N, 11.82; Cl, 15.22.
2-(4-{[1-Methyl-4-(pyridin-4-yl)-1H-pyrazol-3-yl]oxy}butyl)quinoline Dihydrochloride (16b)Compound 16b was prepared from 13 and 15b26) in a manner similar to that described for compound 16e, with a yield of 61% as a pale pink solid. 1H-NMR (DMSO-d6) δ: 1.87–1.98 (2H, m), 2.03–2.14 (2H, m), 3.35 (2H, t, J=7.5 Hz), 3.76 (3H, s), 4.35 (2H, t, J=6.2 Hz), 7.86 (1H, dd, J=7.5, 7.5 Hz), 7.98 (1H, d, J=8.3 Hz), 8.06 (1H, t, J=7.8 Hz), 8.11 (2H, d, J=7.0 Hz), 8.25 (1H, d, J=8.1 Hz), 8.44 (1H, d, J=8.1 Hz), 8.61 (1H, s), 8.73 (2H, d, J=7.0 Hz), 8.96 (1H, br d); MS (ESI) m/z 359 [M+H]+. Anal. Calcd for C22H22N4O·2HCl·3H2O: C, 54.44; H, 6.23; N, 11.54; Cl, 14.61. Found: C, 54.30; H, 5.97; N, 11.51; Cl, 14.71.
N-Methyl-N-(3-{[1-methyl-4-(pyridin-4-yl)-1H-pyrazol-3-yl]oxy}propyl)quinolin-2-amine Dihydrochloride (16c)Compound 16c was prepared from 13 and 15c in a manner similar to that described for compound 16e, with a yield of 23% as an off-white solid. 1H-NMR (DMSO-d6) δ: 2.22–2.32 (2H, m), 3.44 (3H, s), 3.73 (3H, s), 4.02–4.14 (2H, m), 4.46 (2H, t, J=5.9 Hz), 7.47–7.52 (2H, m), 7.73–7.79 (1H, m), 7.88–7.92 (1H, m), 8.01 (2H, d, J=6.6 Hz), 8.16 (1H, d, J=8.3 Hz), 8.35 (1H, d, J=9.8 Hz), 8.55 (1H, s), 8.65 (2H, d, J=6.6 Hz); MS (ESI) m/z 374 [M+H]+. Anal. Calcd for C22H23N5O·2HCl·0.8H2O: C, 57.34; H, 5.82; N, 15.20; Cl, 15.39. Found: C, 57.44; H, 5.76; N, 15.16; Cl, 15.63.
2-[4-({[1-Methyl-4-(pyridin-4-yl)-1H-pyrazol-3-yl]oxy}methyl)piperidin-1-yl]quinoline Dihydrochloride (16d)Compound 16d was prepared from 13 and 15d in a manner similar to that described for compound 16e, with a yield of 56% as a colorless solid. 1H-NMR (DMSO-d6) δ: 1.49–1.61 (2H, m), 2.00–2.08 (2H, m), 2.31–2.43 (1H, m), 3.40–3.50 (2H, m), 3.80 (3H, s), 4.24 (2H, d, J=6.6 Hz), 4.76 (2H, br d J=12.4 Hz), 7.47–7.54 (1H, m), 7.65 (1H, d, J=9.8 Hz), 7.75–7.81 (1H, m), 7.93 (1H, dd, J=8.0, 1.2 Hz), 8.11 (2H, d, J=7.0 Hz), 8.41–8.50 (2H, m), 8.69 (1H, s), 8.73 (2H, d, J=7.0 Hz); MS (ESI) m/z 400 [M+H]+. Anal. Calcd for C24H25N5O·2.2HCl·3.1H2O: C, 53.82; H, 6.29; N, 13.08; Cl, 14.56. Found: C, 53.87; H, 6.28; N, 12.96; Cl, 14.78.
4-(1-Oxidoquinolin-2-yl)butan-1-ol (20b)To a mixture of 15b26) (1.49 g, 7.38 mmol) in CH2Cl2 (30 mL) was added 3-chloroperbenzoic acid (75% purity; 2.21 g, 9.59 mmol), and the mixture was stirred at room temperature for 4 h. The mixture was partitioned between CHCl3 and 1 M NaOH aqueous solution, and the organic layer was dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by silica gel column chromatography (0–3% MeOH in CHCl3) to give 20b (1.38 g, 86%) as a pale yellow solid. 1H-NMR (DMSO-d6) δ: 1.47–1.56 (2H, m), 1.71–1.82 (2H, m), 3.02 (2H, t, J=7.7 Hz), 3.42–3.48 (2H, m), 4.42 (1H, t, J=5.2 Hz), 7.54 (1H, d, J=8.6 Hz), 7.65–7.72 (1H, m), 7.77–7.83 (1H, m), 7.87 (1H, d, J=8.6 Hz), 8.05 (1H, d, J=8.1 Hz), 8.58 (1H, d, J=8.7 Hz); MS (ESI) m/z 218 [M+H]+.
3-(1-Oxidoquinolin-2-yl)propan-1-ol (20a)Compound 20a was prepared from 15a in a manner similar to that described for compound 20b, with a yield of 99% as a brown solid. 1H-NMR (DMSO-d6) δ: 1.84–1.95 (2H, m), 3.06 (2H, t, J=7.5 Hz), 3.45–3.52 (2H, m), 4.65 (1H, t, J=5.3 Hz), 7.54 (1H, d, J=8.6 Hz), 7.66–7.72 (1H, m), 7.78–7.84 (1H, m), 7.88 (1H, d, J=8.6 Hz), 8.05 (1H, d, J=8.1 Hz), 8.58 (1H, d, J=8.6 Hz); MS (ESI) m/z 204 [M+H]+.
2-(4-{[1-Methyl-4-(pyridin-4-yl)-1H-pyrazol-3-yl]oxy}butyl)quinoline 1-Oxide (21b)To a mixture of 13 (930 mg, 5.31 mmol), 20b (1.27 g, 5.84 mmol) and tributylphosphine (2.15 g, 10.6 mmol) in THF (93 mL) was added ADDP (2.68 g, 10.6 mmol), and the mixture was stirred at room temperature for 2 h before the mixture was concentrated in vacuo. The residue was suspended in toluene and the precipitate was removed by filtration. The filtrate was concentrated in vacuo and the residue was purified by silica gel column chromatography (0–50% EtOAc in CHCl3) to give 21b (1.50 g, 76%) as a pale yellow solid. 1H-NMR (DMSO-d6) δ: 1.82–1.98 (4H, m), 3.11 (2H, t, J=7.2 Hz), 3.72 (3H, s), 4.29 (2H, t, J=6.0 Hz), 7.52–7.60 (3H, m), 7.65–7.71 (1H, m), 7.77–7.84 (1H, m), 7.87 (1H, d, J=8.6 Hz), 8.05 (1H, d, J=8.1 Hz), 8.20 (1H, s), 8.40–8.45 (2H, m), 8.57 (1H, d, J=8.6 Hz); MS (ESI) m/z 375 [M+H]+.
2-(3-{[1-Methyl-4-(pyridin-4-yl)-1H-pyrazol-3-yl]oxy}propyl)quinoline 1-Oxide (21a)Compound 21a was prepared from 13 and 20a in a manner similar to that described for compound 21b, with a yield of 78% as a pale yellow solid. 1H-NMR (DMSO-d6) δ: 2.25–2.34 (2H, m), 3.21 (2H, t, J=7.4 Hz), 3.72 (3H, s), 4.33 (2H, t, J=6.2 Hz), 7.45–7.50 (2H, m), 7.57 (1H, d, J=8.6 Hz), 7.65–7.71 (1H, m), 7.78–7.84 (1H, m), 7.85 (1H, d, J=8.6 Hz), 8.00–8.05 (1H, m), 8.20 (1H, s), 8.34 (2H, d, J=5.6 Hz), 8.58 (1H, d, J=8.8 Hz); MS (ESI) m/z 361 [M+H]+.
4-{[1-Methyl-4-(pyridin-4-yl)-1H-pyrazol-3-yl]oxy}-1-(quinolin-2-yl)butyl Acetate (22b)A mixture of 21b (1.40 g, 3.74 mmol) and acetic anhydride (14.2 mL, 150 mmol) was stirred at 80°C for 4 h. After cooling at room temperature, the mixture was concentrated in vacuo. The residue was purified by NH silica gel column chromatography (0–50% EtOAc in CHCl3) to give 22b (902 mg, 58%) as a yellow oil. 1H-NMR (DMSO-d6) δ: 1.82–1.91 (1H, m), 2.15 (3H, s), 3.26–3.32 (4H, m), 3.70 (3H, s), 4.26 (2H, t, J=6.3 Hz), 7.52–7.64 (4H, m), 7.75–7.80 (1H, m), 7.97–8.01 (2H, m, J=9.3), 8.19 (1H, s), 8.38–8.44 (3H, m); MS (ESI) m/z 417 [M+H]+.
3-{[1-Methyl-4-(pyridin-4-yl)-1H-pyrazol-3-yl]oxy}-1-(quinolin-2-yl)propyl Acetate (22a)Compound 22a was prepared from 21a in a manner similar to that described for compound 22b, with a yield of 77% as a yellow oil. 1H-NMR (DMSO-d6) δ: 2.13 (3H, s), 3.27–3.35 (2H, m), 3.71 (3H, s), 4.34–4.40 (2H, m), 6.04–6.10 (1H, m), 7.42–7.46 (2H, m), 7.56–7.63 (2H, m), 7.74–7.80 (1H, m), 7.94–8.03 (2H, m), 8.19 (1H, s), 8.31–8.35 (2H, m), 8.38 (1H, d, J=8.5 Hz); MS (ESI) m/z 403 [M+H]+.
4-{[1-Methyl-4-(pyridin-4-yl)-1H-pyrazol-3-yl]oxy}-1-(quinolin-2-yl)butan-1-ol (23b)To a mixture of 22b (895 mg, 2.15 mmol) in MeOH (13 mL) was added 1 M NaOH aqueous solution (6.45 mL, 6.45 mmol), and the mixture was stirred at room temperature for 3 h. The mixture was partitioned between CHCl3 and brine, and the organic layer was dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by silica gel column chromatography (1–5% MeOH in EtOAc) to give 23b (444 mg, 55%) as a pale yellow solid. 1H-NMR (DMSO-d6) δ: 1.80–2.07 (4H, m), 3.71 (3H, s), 4.25 (2H, t, J=6.0 Hz), 4.80–4.85 (1H, m), 5.63 (1H, d, J=4.8 Hz), 7.53–7.60 (3H, m), 7.70 (1H, d, J=8.5 Hz), 7.71–7.76 (1H, m), 7.94–7.98 (2H, m), 8.19 (1H, s), 8.36 (1H, d, J=8.5 Hz), 8.41–8.44 (2H, m); MS (ESI) m/z 375 [M+H]+. Anal. Calcd for C22H22N4O2·0.1H2O: C, 70.23; H, 5.95; N, 14.89. Found: C, 70.12; H, 5.90; N, 14.79.
3-{[1-Methyl-4-(pyridin-4-yl)-1H-pyrazol-3-yl]oxy}-1-(quinolin-2-yl)propan-1-ol (23a)Compound 23a was prepared from 22a in a manner similar to that described for compound 23b, with a yield of 58% as a pale yellow solid. 1H-NMR (DMSO-d6) δ: 2.17–2.28 (1H, m), 2.35–2.46 (1H, m), 3.72 (3H, s), 4.35–4.45 (2H, m), 4.96–5.02 (1H, m), 5.76 (1H, d, J=4.9 Hz), 7.44–7.48 (2H, m), 7.54–7.60 (1H, m), 7.71–7.77 (2H, m), 7.92–8.00 (2H, m), 8.19 (1H, s), 8.33–8.38 (3H, m); MS (ESI) m/z 361 [M+H]+. Anal. Calcd for C21H20N4O2: C, 69.98; H, 5.59; N, 15.55. Found: C, 69.99; H, 5.61; N, 15.48.
4-{[1-Methyl-4-(pyridin-4-yl)-1H-pyrazol-3-yl]oxy}-1-(quinolin-2-yl)butan-1-one (24)To a solution of 23b (312 mg, 0.83 mmol) in CH2Cl2 (31 mL) was added MnO2 (290 mg, 3.33 mmol), and the mixture was stirred at room temperature for 1 d. The mixture was filtered through celite pad and concentrated in vacuo. The residue was purified by silica gel column chromatography (0–5% MeOH in EtOAc) to give a solid, which was triturated with EtOAc to give 24 (138 mg, 45%) as a colorless solid. 1H-NMR (DMSO-d6) δ: 2.20–2.28 (2H, m), 3.56 (2H, t, J=7.1 Hz), 3.70 (3H, s), 4.36 (2H, t, J=6.3 Hz), 7.52–7.55 (2H, m), 7.73–7.78 (1H, m), 7.85–7.90 (1H, m), 8.04–8.11 (2H, m), 8.13 (1H, d, J=8.5 Hz), 8.19 (1H, s), 8.37–8.41 (2H, m), 8.54 (1H, d, J=8.5 Hz); MS (ESI) m/z 373 [M+H]+. Anal. Calcd for C22H20N4O2·0.35H2O: C, 69.77; H, 5.51; N, 14.79. Found: C, 69.85; H, 5.38; N, 14.68.
2-(1-Fluoro-4-{[1-methyl-4-(pyridin-4-yl)-1H-pyrazol-3-yl]oxy}butyl)quinoline Dihydrochloride (25)Under argon gas atmosphere, to a mixture of 23 (128 mg, 0.34 mmol) in CH2Cl2 (5.1 mL) cooled with dryice-acetone bath was added diethylaminosulfur trifluoride (72 mg, 0.44 mmol), and the mixture was stirred at same temperature for 2 h and at 0°C for another 6 h. The reaction was quenched with water and extracted with CHCl3. The organic layer was washed with saturated NaHCO3 aqueous solution and brine, dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by silica gel column chromatography (0–5% MeOH in EtOAc) to give a colorless oil (28 mg), which was dissolved in EtOAc (3.8 mL) and 4 M HCl/EtOAc (85 µL, 0.34 mmol) was added to the mixture. The mixture was stirred at room temperature for 2 h, and the precipitate was collected by filtration to give 25 (21 mg, 14%) as a pale yellow solid. 1H-NMR (DMSO-d6) δ: 1.95–2.06 (2H, m), 2.19–2.35 (2H, m), 3.77 (3H, s), 4.38 (2H, t, J=6.5 Hz), 5.79–5.97 (1H, m), 7.63–7.69 (1H, m), 7.72 (1H, d, J=8.4 Hz), 7.79–7.84 (1H, m), 8.03 (2H, dd, J=8.7, 8.7 Hz), 8.09 (2H, d, J=7.0 Hz), 8.52 (1H, d, J=8.6 Hz), 8.62 (1H, s), 8.70 (2H, d, J=7.0 Hz); MS (ESI) m/z 377 [M+H]+; HR-MS Calcd for C22H21FN4O [M+H]+ 377.1778. Found 377.1780.
5-(Quinolin-2-yl)thiophene-2-carbaldehyde (27)Compound 27 was prepared from 5-formyl-2-thienylboronic acid (26) and 2-chloroquinoline in a manner similar to that described for compound 11, with a yield of 43% as a beige solid. 1H-NMR (DMSO-d6) δ: 7.61–7.67 (1H, m), 7.78–7.84 (1H, m), 8.00–8.06 (2H, m), 8.12 (1H, d, J=4.0 Hz), 8.20 (1H, d, J=4.0 Hz), 8.26 (1H, d, J=8.6 Hz), 8.51 (1H, d, J=8.6 Hz), 9.99 (1H, s); MS (EI) m/z 239 [M]+.
[5-(Quinolin-2-yl)-2-thienyl]methanol (28)To a mixture of 27 (172 mg, 0.72 mmol) in EtOH (5.2 mL) cooled with ice-water bath was added NaBH4 (27 mg, 0.72 mmol), and the mixture was stirred at same temperature for 2 h. The mixture was partitioned between EtOAc and a 1 : 1 mixture of brine and water, and the organic layer was dried over MgSO4, filtered and concentrated in vacuo to give 28 (171 mg, 99%) as a pale yellow solid. 1H-NMR (DMSO-d6) δ: 4.68 (2H, dd, J=5.7, 0.8 Hz), 5.57 (1H, t, J=5.7 Hz), 7.03–7.06 (1H, m), 7.52–7.58 (1H, m), 7.71–7.77 (1H, m), 7.83 (1H, d, J=3.7 Hz), 7.91–7.96 (2H, m), 8.06 (1H, d, J=8.7 Hz), 8.37 (1H, d, J=8.5 Hz); MS (ESI) m/z 242 [M+H]+.
2-[5-({[1-Methyl-4-(pyridin-4-yl)-1H-pyrazol-3-yl]oxy}methyl)-2-thienyl]quinoline Hemisuccinate (29)To a solution of 28 (320 mg, 1.33 mmol) in CH2Cl2 (6.4 mL) was added SOCl2 (0.29 mL, 3.97 mmol), and the mixture was stirred at room temperature for 3 h before the addition of toluene. The precipitate was collected by filtration to give a beige solid (302 mg). To a mixture of above-obtained beige solid and 13 (179 mg, 1.02 mmol) in DMF (6.0 mL) was added K2CO3 (352 mg, 2.55 mmol), and the mixture was stirred at 60°C for 7 h. After cooling at room temperature, the mixture was partitioned between EtOAc and a 1 : 1 mixture of brine and water, and the aqueous layer was extracted with EtOAc. The combined organic layer was dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by silica gel column chromatography (0–10% MeOH in CHCl3) to give a beige oil (213 mg), which was dissolved in EtOAc (9.0 mL). To the solution was added succinic acid (35 mg), and the mixture was stirred at room temperature for 1 h. The precipitate was collected by filtration to give 29 (205 mg, 39%) as a beige solid. 1H-NMR (DMSO-d6) δ: 2.42 (2H, s), 3.80 (3H, s), 5.55 (2H, s), 7.34 (1H, d, J=3.7 Hz), 7.53–7.61 (3H, m), 7.72–7.79 (1H, m), 7.92 (1H, d, J=3.7 Hz), 7.93–7.99 (2H, m), 8.11 (1H, d, J=8.7 Hz), 8.29 (1H, s), 8.40 (1H, d, J=8.6 Hz), 8.44–8.50 (2H, m), 12.16 (1H, br s); MS (ESI) m/z 399 [M+H]+. Anal. Calcd for C23H18N4OS·0.5C4H6O4: C, 65.63; H, 4.63; N, 12.25; S, 7.01. Found: C, 65.37; H, 4.75; N, 12.16; S, 6.98.
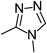
1-Methyl-4-(pyridin-4-yl)-1H-pyrazol-3-amine (31)To a mixture of pyridin-4-ylacetonitrile hydrochloride (30, 2.01 g, 13.0 mmol) in THF (30 mL) and ethanol (30 mL) was added 1 M NaOH aqueous solution (13.0 mL, 13.0 mmol), and the mixture was filtered and the filtrate was concentrated in vacuo. To the residue dissolved in DMF (19 mL) was added DMF-DMA (3.48 mL, 26.0 mmol), and the mixture was stirred at 80°C for 1 h. After cooling at room temperature, the mixture was concentrated in vacuo. To the residue in MeOH (20 mL) were added acetic acid (780 mg, 13.0 mmol) and methylhydrazine (826 µL, 15.6 mmol), and the mixture was stirred at 60°C for 12 h. After cooling at room temperature, the mixture was concentrated in vacuo. The residue was purified by silica gel column chromatography (1–10% MeOH in CHCl3) to give 31 (400 mg, 18%) as an orange solid. 1H-NMR (DMSO-d6) δ: 3.64 (3H, s), 4,90 (2H, s), 7.44–7.47 (2H, m), 7.96 (1H, s), 8.40–8.43 (2H, m); MS (ESI) m/z 175 [M+H]+.
1-Methyl-4-(pyridin-4-yl)-N-[4-(quinolin-2-yl)benzyl]-1H-pyrazol-3-amine (32)To a mixture of 4-(quinolin-2-yl)benzaldehyde27) (284 mg, 1.22 mmol) and 31 (177 mg, 1.02 mmol) in 1,2-dichloroethane (1.0 mL) was added titanium(IV) isopropoxide (0.45 mL, 1.53 mmol), and the mixture was stirred at 85°C for 2 h. To the resultant mixture cooled with ice-water bath were added MeOH (5 mL) and NaBH4 (270 mg, 7.14 mmol), and the mixture was stirred at room temperature for 5 h. The reaction was quenched with saturated NaHCO3 aqueous solution and diluted with CHCl3. The mixture was filtered through celite pad and the organic layer of the filtrate was washed with brine, dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by silica gel column chromatography (1–7% MeOH in CHCl3) to give 32 (138 mg, 35%) as a pale yellow solid. 1H-NMR (DMSO-d6) δ: 3.67 (3H, s), 4.46 (2H, d, J=6.0 Hz), 5.95 (1H, t, J=6.0 Hz), 7.50–7.53 (2H, m), 7.55–7.61 (3H, m), 7.75–7.80 (1H, m), 7.97–8.01 (2H, m), 8.06 (1H, d, J=8.4 Hz), 8.13 (1H, d, J=8.7 Hz), 8.20–8.24 (2H, m), 8.42–8.47 (3H, m); MS (ESI) m/z 392 [M+H]+. Anal. Calcd for C25H21N5·1.3H2O·0.1CHCl3: C, 70.63; H, 5.60; N, 16.41. Found: C, 70.90; H, 5.37; N, 16.17.
N,1-Dimethyl-4-(pyridin-4-yl)-N-[4-(quinolin-2-yl)benzyl]-1H-pyrazol-3-amine Dihydrochloride (33)To a solution of 32 (77 mg, 0.20 mmol) and acetic acid (0.80 mL) in CH2Cl2 (4.0 mL) was added 36% formaldehyde aqueous solution (150 µL, 1.96 mmol), and the mixture was stirred at room temperature for 10 min. To the resultant mixture was added sodium triacetoxyborohydride (167 mg, 0.79 mmol), and the mixture was stirred at room temperature for 2 h. To the resultant mixture were added 36% formaldehyde aqueous solution (150 µL, 1.96 mmol) and sodium triacetoxyborohydride (167 mg, 0.79 mmol), and the mixture was stirred at room temperature overnight. To the resultant mixture were added 36% formaldehyde aqueous solution (150 µL, 1.96 mmol) and sodium triacetoxyborohydride (167 mg, 0.79 mmol) again, and the mixture was stirred at room temperature for 12 h. The reaction was quenched with saturated NaHCO3 aqueous solution and extracted with CHCl3. The organic layer was dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by silica gel column chromatography (1–7% MeOH in CHCl3) to give a pale yellow amorphous, which was dissolved in MeOH (5 mL) and 4 M HCl/EtOAc (0.15 mL, 0.60 mmol) was added to the mixture. The mixture was concentrated in vacuo, and the residue was washed with diisopropylether/2-propanol to give a yellow solid. 1H-NMR (DMSO-d6) δ: 2.68 (3H, s), 3.82 (3H, s), 4.32 (2H, s), 7.54 (2H, d, J=8.3 Hz), 7.65–7.70 (1H, m), 7.83–7.89 (1H, m), 8.08 (1H, d, J=8.0 Hz), 8.17–8.27 (6H, m), 8.58–8.63 (m, 2H), 8.78 (2H, d, J=7.0 Hz); MS (ESI) m/z 406 [M+H]+. Anal. Calcd for C26H23N5·2HCl·2.5H2O·0.75C3H8O: C, 59.68; H, 6.38; N, 12.32; Cl, 12.47. Found: C, 59.91; H, 6.42; N, 12.18; Cl, 12.24.
2-[4-(Chloromethyl)phenyl]quinoline Hydrochloride (34)To a mixture of 11 (500 mg, 2.13 mmol) in CH2Cl2 (7.5 mL) was added SOCl2 (758 mg, 6.37 mmol), and the mixture was stirred at r.t for 2 h. To the reaction mixture was added toluene and the precipitate was collected by filtration to give 2-[4-(chloromethyl)phenyl]quinoline hydrochloride (616 mg, quant) as a colorless solid. 1H-NMR (DMSO-d6) δ: 4.88 (2H, s), 7.64–7.71 (3H, m), 7.85–7.90 (1H, m), 8.07–8.11 (1H, m), 8.19 (1H, d, J=8.5 Hz), 8.23 (1H, d, J=8.7 Hz), 8.27–8.31 (2H, m), 8.63 (1H, d, J=8.7 Hz); MS (ESI) m/z 254, 256 [M+H]+.
4-(Pyridin-4-yl)-1H-pyrazol-3-ol (35)Compound 35 was prepared from 12 and hydrazine hydrate in a manner similar to that described for compound 13, with a yield of 80% as a pink solid. 1H-NMR (DMSO-d6) δ: 7.65 (2H, s), 8.12 (1H, s), 8.38 (2H, s), 10.6 (1H, br s), 12.1 (1H, br s); MS (ESI) m/z 162 [M+H]+.
1-[3-Hydroxy-4-(pyridin-4-yl)-1H-pyrazol-1-yl]ethanone (36)To a mixture of 35 (7.84 g, 48.6 mmol) in pyridine (78 mL) was added Ac2O (4.81 mL, 50.9 mmol), and the mixture was stirred at 100°C for 2 h before the mixture was concentrated in vacuo. The residue was washed with water and hexane to give 36 (9.89 g, quant) as yellow solid. 1H-NMR (DMSO-d6) δ: 2.55 (3H, s), 7.80–7.84 (2H, m), 8.53–8.56 (2H, m), 8.92 (1H, s), 12.0 (1H, br s); MS (ESI) m/z 204 [M+H]+.
2-[4-({[4-(Pyridin-4-yl)-1-(2,2,2-trifluoroethyl)-1H-pyrazol-3-yl]oxy}methyl)phenyl]quinoline Hydrochloride (37)To a mixture of 36 (2.03 g, 10.0 mmol), 34 (3.19 g, 11.0 mmol) in DMF (77 mL) was added K2CO3 (4.15 g, 30.0 mmol), and the mixture was stirred at 60°C for 3 h before the mixture was concentrated in vacuo. To the residue were added MeOH (80 mL) and water (20 mL), and the mixture was stirred at 60°C for 3 h before the mixture was concentrated in vacuo. The residue was washed with water and purified by silica gel column chromatography (0–5% MeOH in CHCl3) to give a yellow solid (1.52 g). To a mixture of the yellow solid obtained above (568 mg) and 1,1,1-trifluoro-2-iodomethane (630 mg, 3.00 mmol) in DMF (20 mL) was added Cs2CO3 (1.47 g, 4.50 mmol), and the mixture was stirred at 60°C for 5 h before the mixture was concentrated in vacuo. The residue was partitioned between water and EtOAc, and the organic layer was concentrated in vacuo. The residue was purified by silica gel column chromatography (1–7% MeOH in CHCl3) to give free form of the title compound, which was dissolved in EtOAc (20 mL). To the mixture was added 4 M HCl/EtOAc (1.5 mL) and the mixture was stirred at r.t. for 30 min. The precipitate was collected by filtration and washed with EtOAc to give 37 (650 mg, 27%) as a pale yellow solid. 1H-NMR (DMSO-d6) δ: 5.19 (2H, q, J=8.9 Hz), 5.54 (2H, s), 7.69–7.75 (1H, m), 7.78 (2H, d, J=8.4 Hz), 7.89–7.94 (1H, m), 8.13 (1H, d, J=7.8 Hz), 8.23–8.38 (6H, m), 8.71 (1H, d, J=8.7 Hz), 8.79–8.84 (2H, m), 8.89 (1H, s); MS (ESI) m/z 461 [M+H]+. Anal. Calcd for C26H19F3N4·2.2HCl·0.3C4H8O2·2.2H2O: C, 53.84; H, 4.65; N, 9.23; Cl, 12.86; F, 9.39. Found: C, 54.00; H, 4.61; N, 9.48; Cl, 13.05; F, 8.81.
Molecular ModelingThe geometry of MP-10 binding to PDE10A enzyme was derived from PDB (code: 3HR1). Structure alignment of MP-10, 16a, and b was performed with the Flexible Alignment tool in the MOE program29) with the geometry of MP-10 as a template and the MMFF94x force field.
Calculation of Atomic Charge DistributionMOE was used to build the ligand structures of MP-10 and 14.33) Conformation search of the ligands were done with Conformation Import module in MOE with MMFF94x force field. Potential energy of each conformation were calculated with PM6 method implemented in MOPAC2012. The lowest energy was selected as energy-minimized conformation to calculate partial charge. The electrostatic potential-fitted atomic partial charges were calculated with the AM1 method implemented in the MOPAC7.1.
PDE10A Enzyme Assay ProtocolThe full-length human PDE10A2 was amplified via PCR using the 1st strand cDNA synthesized from the total RNA isolated from human neuroblastoma TGW cell line. The PCR products were cloned into a pCR2.1-TOPO vector (Invitrogen Inc.) to confirm sequences. The confirmed plasmid was digested with restricted enzymes, BamHI/HindIII, and this digested product was inserted into a pFastBac1 vector (Invitrogen Inc.). Human PDE10A2 enzyme protein was expressed in a Spodoptera frugiperda Sf9 insect cell using the Bac-to-Bac Baculovirus Expression System (Invitrogen Inc.). The infected Sf9 cells were collected via centrifuge and the medium was removed. The collected cells were lysed by sonication in lysis buffer (50 mM Tris–HCl [pH 8.0], 150 mM NaCl, 3 mM DTT, 0.1% NP-40, and 20% glycerol with protease inhibitors), after which the lysate was centrifuged and supernatant was collected to obtain PDE10A2 enzyme solution. We confirmed the PDE10A2 expression via Western blot analysis. Inhibitory effects of compounds on human PDE10A enzyme activity was assessed by measuring the amount of cAMP via the Homogeneous Time-Resolved Fluorescence (HTRF) detection method. The assay was performed in 12 µL samples containing a optimal amount of the PDE10A enzyme, buffer (40 mM Tris–HCl pH 7.5; 5 mM MgCl2), 0.1 µM cAMP and various concentrations of compounds (0.1 nM to 10 µM). After compounds were preincubated for 30 min with the enzyme, the reaction was initiated by adding the substrate cAMP and the mixture was incubated for 60 min at room temperature with agitation. The reaction was terminated by the addition of the fluorescence acceptor (cAMP labeled with the dye d2) and the fluorescence donor (anti-cAMP antibody labeled with Cryptate, Cisbio). After 60 min, the fluorescence transfer corresponding to the amount of residual cAMP was measured at lex. 320 nm, lem. 620 nm and lem. 665 nm using an Envision plate reader (PerkinElmer, Inc.) and signal ratio (665/620) was calculated. The ratio determined in the absence of enzyme was subtracted from all data. The obtained results were converted to activity relative to an uninhibited control (100%) and IC50 values were calculated using Prism software (GraphPad Software, Inc.).
In Vitro Enzyme Assays for Profiling PDE SelectivityPhosphodiesterases 4B, D, 9A, and 10A were generated from full-length human recombinant clones. PDE2A was isolated from rat. while PDE3 and 5 were isolated from rabbit. PDE activity was measured with the preferred substrates using a scintillation proximity assay. For PDE2A, 3, 4B, D, 10A, cAMP was used, and for PDE5, and 9A, cGMP was used. The effect of PDE inhibitors was investigated by assaying a fixed amount of enzyme and varying inhibitor concentrations. For determination of IC50 values, the Hill-plot two-parameter model was used.
In Vivo Behavioral Assay in MiceTwo behavioral tests described below were conducted for mice. All animal experimental procedures were approved by the Institutional Animal Care and Use Committee of Astellas Pharma Inc. Further, the Astellas Pharma Inc. Tsukuba Research Center was awarded Accreditation Status by the AAALAC International. All efforts were made to minimize the number of animals used and to avoid suffering and distress.
Phencyclidine-Induced Hyperlocomotion: ICR mice aged 5–6 weeks were used to evaluate the effect of PDE10A inhibitor on hyper-locomotion induced by the N-methyl-D-aspartate (NMDA) antagonist phencyclidine (PCP). Immediately after oral administration of either vehicle or agent as pre-treatment, mice were placed into individual plastic test cages (30×35×17.5 cm) of a SUPERMEX system (PAT.P; Muromachi Kikai Co., Ltd.), and measurement of locomotor activity was started. After 1 h, the mice were injected with a post-treatment of saline or PCP (2.5 mg/10 mL/kg, subcutaneously (s.c.)), and locomotor activity was measured for a further 60 min. Total locomotor activity for 60 min post-treatment was calculated.
NORT in Neonatally PCP-Treated Mice: Three-day-old male ddY mice were housed 10–12 per cage with a stepmother. Saline or PCP (15 mg/kg) was administered subcutaneously once daily on days 7, 9, and 11 after birth. The mice were separated from their mother at 3 weeks of age, and used for NORT at 8–9 weeks old. Neonatal mice were treated with PCP, and the NORT was conducted as previously described.37)
In Vitro Metabolic Pathway in Mouse Liver MicrosomesThe incubation mixture consisted of the following: 10 µM MP-10, 100 mM phosphate buffer (pH 7.4), liver microsomes (0.2 mg protein), and a reduced nicotinamide adenine dinucleotide phosphate (NADPH)-generating system (15 mM glucose 6-phosphate, 3 mM NADP+, 5 mM magnesium chloride, and 0.3 U glucose 6-phosphate dehydrogenase) in a total volume of 500 µL. The mixture was preincubated for 5 min at 37°C. A reaction was started by the addition of MP-10 (added in acetonitrile–water, 50 : 50, v/v; final solvent concentration not exceeding 0.5% [v/v]). The samples were incubated at 37°C for 15 and 60 min, and the reaction was stopped by the addition of 1 mL cold MeCN. The reaction mixture was centrifuged at 16000×g for 5 min, and the supernatant was evaporated under a steam of nitrogen gas at 40°C. The dried extracts containing the parent drug and metabolites were dissolved in 200 µL of acetonitrile–water (50 : 50, v/v), and 800 µL of acetonitrile was added. Centrifugation and evaporation of the supernatant was repeated once again. The dried extracts were then dissolved in 200 µL of MeCN–water (50 : 50, v/v), and filtered through a membrane filter, and 5 µL of the filtrate was analyzed using LC-MS/MS with Waters Ultra Performance Liquid Chromatography (UPLC) system and Thermo Scientific LTQ-Orbitrap mass spectrometer.
Mouse and Human Liver Microsomal AssaysPooled mouse or human liver microsomes (Xenotech LLC.) were diluted in 100 mM KH2PO4/K2HPO4 buffer (pH 7.4) containing 0.1 mM ethylenediaminetetraacetic acid (EDTA). The incubation mixtures (270 µL total volume), which contained 0.2 mg/mL of microsomal proteins, and 1 mM NADPH (30 µL) were pre-incubated for 5 min at 37°C. Reactions were initiated by the addition of 0.2 µM of substrates. After the appropriate incubation time (0, 15, 30, 45 min), 50 µL of incubation mixture was transferred into 80% acetonitrile containing internal standard (50 ng/mL diazepam, 250 µL) and centrifuged for 10 min at 2800 rpm. The supernatant (200 µL) was prepared and analyzed via LC-MS/MS with a Surveyor HPCL system (Thermo Fisher Scientific Inc.) and TSQ Quantum ultra tandem triple quadrupole mass spectrometer (Thermo Fisher Scientific Inc.). The in vitro intrinsic clearance (CLint, vitro) was calculated using Eq. 1, which is based on the time course of the residual ratio of the compounds.38)
 | (1) |
where
Ke is the disappearance rate constant.
Mouse Pharmacokinetic StudyCompound 14 or TP-10 was administered to ICR mice as a 30 mg/kg solution of saline containing 5% dimethylsulfoxide (DMSO) and 5% Cremophor. Blood samples were collected using syringes containing heparin sodium at 0.5, 1, 2, and 4 h after intraperitoneal administration. Blood samples were kept on ice and centrifuged within 0.5 h of collection at 16000×g, for 2 min at 4°C to obtain plasma, which was then stored at −20°C prior to analysis. Whole brain samples were also removed from the same animals as blood samples just after the collection of blood samples,and stored at −20°C and homogenized in 4-fold volume of phosphate buffered saline (pH 7.4) before extraction processing. Extraction and analysis of compound concentrations were performed via LC-MS/MS with a Prominence HPLC (Shimadzu Corporation) and API4000 Triple Quadrupole Mass Spectrometer (AB SCIEX).