Abstract
Although cytochromes P450 2C9 (CYP2C9) and 2C19 (CYP2C19) have 91% amino acid identity, they have different substrate specificities. Previous studies have suggested that several amino acid residues may be involved in substrate specificity. In this study, we focused on the roles of two amino acids, residues 72 and 241. The amino acids in these positions have opposite charges in CYP2C9 and 2C19; the former has lysines in both positions (Lys72 and Lys241), and the latter has glutamic acids (Glu72 and Glu241). Reciprocal mutants for both CYP2C19 and 2C9 were produced, and their metabolic activities and spectroscopic properties were examined using three tricyclic antidepressant (TCA) drugs: amitriptyline, imipramine, and dothiepin. Although CYP2C19 wild-type (WT) had a high metabolic activity for all three drugs, the E72K mutation decreased enzymatic activity by 29–37%, while binding affinities were diminished 2.5- to 20-fold. On the other hand, low activity and low affinity of CYP2C9 WT were recovered notably by K72E mutation. The metabolic activities and binding affinities were minimally affected by CYP2C19 E241K and CYP2C9 K241E mutations. We could also show linear correlations between metabolic activities and binding affinities, and hence we conclude that amino acid residue 72 plays a key role in TCA drug metabolism by limiting the binding affinities of CYP2C19 and CYP2C9.
Depression is a psychiatric condition that affects 120 million people worldwide, and can interfere with independence and productivity in essentially all aspects of daily life. Depression is also associated with a high risk of self-harm, and ultimately suicide. Despite the considerable advances in depression treatments, tricyclic antidepressants (TCAs) remain an important therapeutic option for depression. TCAs such as amitriptyline (AMI), imipramine (IMI), and dothiepin (DOT) are heterocyclic chemical compounds (Fig. 1). These drugs are characterized by substantial pre-systemic first-pass metabolism and their clearance is dependent primarily on hepatic cytochrome P450 (CYP) oxidative enzymes, in particular CYP2C19.1) Because the activity of some CYP isoforms is individually influenced by genetic variations, there is wide inter-individual variability in TCA pharmacokinetics and active metabolite production,2,3) which complicates the clinical use of these drugs. Therefore, the understanding of TCA drug metabolism at the molecular level is essential for the safe use of these drugs.

Fig. 1. Chemical Structures of AMI, IMI, and DOT, Classified as Tricyclic Antidepressant Drugs
CYPs are a superfamily of heme-containing enzymes that are mainly responsible for oxidative transformation of xenobiotics in humans, animals, and plants. The CYP1, 2, and 3 families in mammals have a wide range of overlapping substrates and play a role in the metabolism of both drug and endogenous substrates through N-, O-, and S-dealkylation, aromatic and aliphatic hydroxylation, epoxidation; oxidative desulfurization, and sulfoxidation.4,5) The CYP2C subfamily includes the CYP2C8, 2C9, 2C18, and 2C19 isoforms in humans.6) Among these isoforms, CYP2C9 and 2C19 are the most conserved and metabolize numerous clinically important drugs.7) CYP2C19 and 2C9 share 91% amino acid to be identical with only 43 amino acid differences in a total of 490 residues (Fig. 2). In spite of the high homology, different substrate specificities have been reported.6) CYP2C19 is responsible for hydroxylation of (S)-mephenytoin and is highly selective for proton pump inhibitors, such as omeprazole and lansoprazole, while the structurally related CYP2C9 has low affinity for these substrates. On the other hand, both CYP2C19 and 2C9 metabolize tolbutamide in a similar manner.8–10)

Studies of chimeric constructs and amino acid substitutions between CYP2C19 and 2C9 have identified several amino acid residues as key determinants for omeprazole 5-hydroxylase activity,11,12) (S)-mephenytoin 4′-hydroxylation,13,14) and binding and metabolism of warfarin,15) diclofenac, and ibuprofen.16,17) It is noteworthy that the role of CYP2C19 residues glutamic acid 72 (Glu72) and glutamic acid 241 (Glu241), and CYP2C9 residues lysine 72 (Lys72) and lysine 241 (Lys241) in metabolic activity and binding affinity for TCA drugs, as well as basic drugs, has not been investigated.
Although a previous study on diclofenac and ibuprofen binding affinity indicated that neither arginine 97 (Arg97) nor Lys72 CYP2C9 residue plays an important role in determining substrate specificity,17) other studies reported that substitution of Arg97 with alanine decreased the diclofenac 4′-hydroxylase activity of CYP2C9 by increasing the Km value.18) Furthermore, another study19) based on different homology models proposed that CYP2C9 Lys72 is positioned within the substrate access channel and may be responsible for substrate specificity.
In this study, we aimed to assess the role of CYP2C19 Glu72 and Glu241, and CYP2C9 Lys72 and Lys241 in enzymatic activities for AMI, IMI, and DOT. CYP2C19 Glu72 and Glu241 were individually mutated to Lys to mimic CYP2C9, and vice versa for CYP2C9. The effects of the mutations were studied in comparison with metabolic activities and binding affinities of wild-type (WT) CYP2C19 and 2C9 on the TCA drugs.
Experimental
Chemicals and ReagentsDOT and cytochrome b5 were purchased from Sigma-Aldrich (St. Louis, MO, U.S.A.). AMI, IMI, and other chemicals were purchased from Wako Pure Chemical Industries, Ltd. (Osaka, Japan). Recombinant human CYP reductase (CPR) was prepared, and the purity and activity of CPR were confirmed according to the reported methods.5,20,21)
Protein Expression and PurificationThe CYP2C19 WT gene was constructed previously, and the CYP2C9 WT gene was designed, synthesized, and ligated into a pET3a-based expression vector (pBEX).22,23) Subsequently, the CYP2C19 E72K, 2C19 E241K, 2C9 K72E, or 2C9 K241E mutations were introduced using the QuikChange (Stratagene) site-directed mutagenesis kit, and the mutated sequences were confirmed by DNA sequencing. Escherichia coli strain BL21 Gold (DE3) was transformed with the plasmids, and WTs and mutants of CYP2C9 and 2C19 were expressed and purified as previously reported,1) with slight modifications for CYP2C9. In order to stabilize the enzyme, 500 µM diclofenac was added to the bacterial cultures after 6 h of cultivation, and cells were homogenized in a high-pressure homogenizer (EmulsiFlex-C5, AVESTIN). Finally, CYP2C9 and 2C19 purities were evaluated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). All CYP preparations showed single bands in the gel as previously reported for CYP2C19 WT,1) and were highly pure for evaluations of metabolic activity and drug binding affinity.
Metabolic ActivityThe enzyme kinetic parameters (Km, Vmax, and CLint) for TCA drug metabolism were measured using the purified WTs and mutants at various substrate concentrations as described previously,1) except for using purified CPR. The concentration and activity of the purified CPR were assessed with a spectrophotometer and with the UPLC system, respectively, and 0.4 µM CPR was added to the reaction mixture.
Drug Binding AffinityThe binding affinities and spectral changes of CYP preparations for AMI, IMI, and DOT were determined by measuring the drug dissociation constants (Kd) by using a spectral titration method. A solution containing 8 µM CYPs was prepared in 100 mM KPi buffer (100 mM potassium phosphate, 0.1 mM ethylenediaminetetraacetic acid, and 20% glycerol, pH 7.4). Stock solutions of drugs were prepared in the same buffer. Small aliquots of the drug stock solution were added with a syringe to the protein solution in a cuvette. The spectrum was scanned at suitable time intervals after drug addition, and the titration was continued until spectral saturation was achieved. The absorbance changes in the Soret band region were analyzed with a UV-visible spectrophotometer (DU-800, Beckman-Coulter).
Results
Kinetics Parameters of EnzymeThe kinetic parameters of enzyme, Km and Vmax, were evaluated with the Michaelis–Menten equation, and the intrinsic clearance (CLint) value, an index of metabolic efficacy, was derived from the Vmax to Km ratio.1)
First, we estimated the enzymatic kinetic parameters of TCA drugs metabolism by CYP2C19 and 2C9 WTs. Our previous study on AMI and DOT suggested that CYP2C19 WT can metabolize TCA drugs, and the Km value of 107 µM for IMI was similar to those for AMI and DOT, also the Vmax was 15 µM/min/µM of CYP, and the CLint was 140 mL/min/µmol of CYP, which was similar to that for DOT (126 mL/min/µmol of CYP). However, the structurally related CYP2C9 WT had lower metabolic activity than CYP2C19 WT. The Km values for AMI, IMI, and DOT were 198, 354, and 253 µM, respectively, and the Vmax values were lower than those for CYP2C19. The CLint value of 31 mL/min/µmol of CYP for all three drugs was also lower (Fig. 3 and Table 1).
Next, we estimated the AMI, IMI, and DOT metabolism kinetic parameters for CYP2C19 E72K and E241K mutants, and 2C9 K72E and K241E mutants (Fig. 3). Remarkably, the CYP2C19 E72K mutant had significantly higher Km values for AMI, IMI, and DOT (98, 139, 108 µM, respectively), with a minimal decrease in the Vmax for each TCA drug (14, 14, 9.3 µM/min/μM of CYP, respectively). These values resulted in an overall decrease in CLint (143, 100, and 86 mL/min/µmol of CYP for AMI, IMI, and DOT, respectively (Table 1). While, CYP2C19 E241K mutation had little effect on both Km and Vmax, and hence, metabolism rates for AMI, IMI, and DOT were slightly affected with CLint values of 192, 135, and 120 mL/min/µmol of CYP, respectively.

Table 1. The Enzymatic Parameters for the Metabolism of AMI, IMI, and DOT by WTs and the Mutants of CYP2C19 and 2C9
| CYPs | AMI | IMI | DOT |
|---|
| Km | Vmax | CLint | Km | Vmax | CLint | Km | Vmax | CLint |
|---|
| 2C19 WT | 69±6 | 15±0.4 | 224±13 | 107±11 | 15±0.6 | 140±9 | 79±9 | 9.9±0.4 | 126±10 |
| 2C19 E72K | 98±3** | 14±0.1 | 143±3** | 139±9* | 14±0.3 | 100±4** | 108±6** | 9.3±0.2 | 86±6** |
| 2C19 E241K | 78±5 | 15±0.3 | 192±9 | 111±9 | 15±0.4 | 135±7 | 82±4 | 9.8±0.1 | 120±4 |
| 2C9 WT | 198±19 | 6.1±0.3 | 31±2 | 354±73 | 11±1.2 | 31±3 | 253±33 | 7.8±0.5 | 31±3 |
| 2C9 K72E | 121±5** | 8.9±0.1 | 74±2** | 298±53 | 13±1.2 | 44±4** | 168±20* | 8.2±0.4 | 49±2** |
| 2C9 K241E | 189±18 | 6.2±0.3 | 33±2 | 332±24 | 11±0.4 | 33±1 | 229±19 | 7.9±0.3 | 34±2 |
The Michaelis constants, Km (µM), and the maximum velocities, Vmax (µM/min/µM of CYP), were provided by fitting of theoretical curves, and the values of intrinsic clearance, CLint (mL/min/µmol of CYP), were calculated with the Km and Vmax values. These values were analyzed statistically, and marked with asterisks (*p<0.05 or **p<0.01) when they were different significantly from those of CYP2C19 WT or CYP2C9 WT.
On the other hand, the K72E mutation increased the metabolic activity of CYP2C9 by decreasing Km to 121, 298, and 168 µM, and slightly increasing Vmax to 8.9, 13, and 8.2 µM/min/μM of CYP for AMI, IMI, and DOT, respectively. Consequently, the CLint values for CYP2C9 K72E were significantly higher than those for CYP2C9 WT. The position of the mutated residue was identical between CYP2C19 and 2C9, implying that the residue may be important for TCA metabolism. However, the K241E mutation did not affect CYP2C9 metabolism of AMI, IMI, and DOT as the CLint values were 33, 33, and 34 mL/min/µmol of CYP, respectively. The measured enzymatic kinetic parameters (Km and Vmax) and the estimated CLint values for CYP2C19 and 2C9 WTs and mutants are summarized in Table 1.
TCA BindingSince the metabolic activity results suggested that mutations at residue 72 could affect TCA binding affinity, we measured the absorption spectra of CYP2C19 and 2C9 WTs and mutants to characterize the type of binding and affinities for TCA drugs. Spectral changes are one of the tools to identify enzyme-bound chemicals as either substrates or inhibitors.24,25)
In this study, we observed type I and reverse type I spectral changes, and CYP2C19 WT with IMI was representative of type I spectral change (Fig. 4A). Type I spectral changes were also observed for CYP2C19 with AMI and DOT, accompanied by high binding affinities (Kd values of 4.0, 17 µM, respectively). Subsequently, the binding affinities and spectral changes of CYP2C19 mutants were also evaluated (Table 2). Remarkably, spectral change was disturbed by the mutations, and both CYP2C19 E72K and E241K mutants showed reverse type I change (Fig. 4B). The binding affinities for the studied TCA drugs were, however, distinct between E72K and E241K mutants; the effect was greater by E72K mutation than by E241K mutation. Typically, the Kd value of CYP2C19 WT for IMI (57 µM) is close to that of E241K (69 µM) but is greatly raised by E72K mutation (144 µM). These results suggested that Glu72 in CYP2C19 plays a greater role than Glu241 in regulating drug binding affinity, although both residues affect drug binding mode. Subsequently, we also evaluated CYP2C9 WT and the K72E and K241E mutants (Table 2). While CYP2C9 WT had low affinity for AMI, IMI, and DOT (301, 354, 278 µM, respectively), the binding affinity was recovered by K72E mutation with Kd values of 103, 283, and 140 µM for AMI, IMI, and DOT, respectively, but was minimally affected by K241E mutation. Both CYP2C9 K72E and K241E mutants, however, showed type I change similarly for all investigated TCA drugs, whereas CYP2C9 WT showed reverse type I. Thus, we could indicate again that residue 72 plays a greater role than residue 241 in regulating TCA binding affinity, although both residues affect drug binding mode.
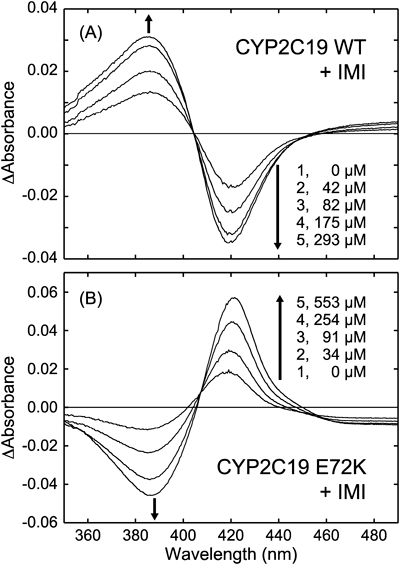
Table 2. Dissociation Constants and Spectral Changes of WTs and the Mutants of CYP2C19 and 2C9 for AMI, IMI, and DOT
| CYPs | AMI | IMI | DOT |
|---|
| Kd (μM) | Spectral changes | Kd (μM) | Spectral changes | Kd (μM) | Spectral changes |
|---|
| 2C19 WT | 4.0±0.4 | Type I | 57±1 | Type I | 17±1 | Type I |
| 2C19 E72K | 79±10 | Reverse Type I | 144±11 | Reverse Type I | 50±1 | Reverse Type I |
| 2C19 E241K | 23±1 | Reverse Type I | 69±4 | Reverse Type I | 25±1 | Reverse Type I |
| 2C9 WT | 301±21 | Reverse Type I | 354±25 | Reverse Type I | 278±14 | Reverse Type I |
| 2C9 K72E | 103±16 | Type I | 283±16 | Type I | 140±9 | Type I |
| 2C9 K241E | 293±22 | Type I | 338±19 | Type I | 248±32 | Type I |
The dissociation constants, Kd (µM), were evaluated by monitoring absorbance changes with drug titration. The patterns of spectral changes were also assigned to be either Type I or reverse Type I.
Discussion
Although the amino acid composition of CYP2C9 and 2C19 is 91% identical, they have different metabolic activities. There are several reports on the critical amino acid residues in either CYP2C9 or 2C19, which are responsible for substrate specificity. Studies with chimeric constructs and amino acid substitutions between CYP2C19 and CYP2C9 found that 2C19 residues 99, 220, and 221 could be the key determinants of omeprazole 5-hydroxylase activity.11) Further, residues 237 and 286 were suggested to play important roles in (S)-mephenytoin 4′-hydroxylation,12,14) and replacement of asparagine 286 with serine (N286S) enhanced tolbutamide p-methyl hydroxylase, diclofenac 4′-hydroxylase, and ibuprofen hydroxylase activities.16) Moreover, the CYP2C19 KSN triple mutant (E241K/N286S/I289N) was reported to metabolize both (R)- and (S)-warfarin with a broadened regioselectivity.15) These studies suggested that the F-G loop might be a part of the substrate access channel in both CYP2C9 and CYP2C19. In this study, we examined the metabolic activities of CYP2C19 and 2C9 for three TCA drugs (AMI, IMI, and DOT). A previous in vitro study indicated that AMI was metabolized primarily by CYP2C19 and 2D6, moderately by CYP1A2 and 2C9, and slightly by CYP3A4.26) Consistent with this, we observed ca. 7-fold higher CLint value for CYP2C19 WT (224 mL/min/µmol of CYP) than for CYP2C9 WT (31 mL/min/µmol of CYP). Further, IMI and DOT were also metabolized by CYP2C9, which is a novel observation.
As shown in Table 1, the Vmax values were minimally disturbed by the mutations for all investigated TCA drugs. However, the Km values were affected by mutations at both positions 72 and 241. Remarkably, CYP2C19 E72K mutant showed higher Km values than CYP2C19 WT, reducing CLint values. On the other hand, low CLint values of CYP2C9 WT were slightly recovered by replacing Lys with Glu at position 72 (K72E) but was minimally at position 241 (K241E). These imply that Glu72 could be more effective than Glu241 for binding of TCA drugs.
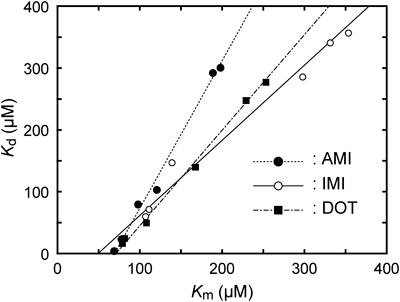
In order to assess the roles of these residues, we examined the correlation between metabolic activity and binding affinity for the TCA drugs. In Fig. 5, dissociation constants (Kd) were plotted against Michaelis constants (Km). Interestingly, the Km and Kd values were linearly correlated for each drug that Glu72 could be important for drug and mainly control the metabolic reactions in both CYP2C19 and 2C9. In addition, the correlation lines were close to each other, implying that properties of TCA drugs could affect either the slope or intercept, and these parameters could be used as indicators to identify the effects of the studied drugs.
Next, we addressed absorption spectral changes in two isoforms. CYP2C19 WT was characterized by type I spectral changes, and both E72K and E241K mutations altered the spectral changes to reverse type I. On the other hand, reverse type I change in CYP2C9 WT was altered to be type I by both K72E and K241E mutations. These suggested that both 72 and 241 residues contribute to the binding mode of TCA drugs. The heme iron in CYPs has either 6- or 5-coordinated state in equilibrium with or without water axial ligand. Reverse type I spectral change indicates that the heme iron prefers the 6-coordinated state by drug binding. This implies that values of Vmax may decrease, because 6-coordinated heme has relatively low redox potential and is difficult to receive electrons from CPR. However, the values of Vmax were altered minimally, even though spectral types were disturbed by mutations in both CYP2C19 and 2C9. In this study, absorbance change was relatively small even in the presence of saturating amount of drugs (ΔA ca. 0.05) (Fig. 3). It implies that the disturbance of heme coordination was too small to affect Vmax values. This remote effect, posed by both 72 and 241 residues, may have resulted in the changes of spectral type as well as binding affinity.
To interpret the effects of the mutations, we superimposed the reported structures of CYP2C9 (pdb code: 1R9O) and CYP2C19 (pdb code: 4GQS). As indicated in ball-and-stick models, residues 72 and 241 were at similar positions in CYP2C19 and 2C9 (Fig. 6). Indeed Glu72 can form a hydrogen bond with histidine 99 (His99) being on a loop between B’- and C-helices (B’C-loop),27) and hence we depicted the notable residues in the B’C-loop as stick models. Remarkably, the structures of CYP2C19 and 2C9 were virtually identical except for some loops, such as the B’C-loop. The difference could be induced by the reciprocal residues at position 72, which affected the orientation of some residues located in the loop. Here, we focused on bulky residues, three phenylalanines, Phe100, Phe110, and Phe114 because of their proximity to the heme. These residues face the active site, and Phe114 was suggested to interact with 0VX, an inhibitor of CYP2C19, by π-stacking in the aromatic rings. Moreover, the different orientations of the corresponding phenylalanines in CYP1B1 and 2A13, were shown to cause alterations in spectral types.25) In this study, all investigated drugs also contained tricyclic parts with aromatic rings, suggesting that they could interact with Phe114 and/or Phe100 by π-stacking. Actually, the location of Phe114 was different in the reported structures of CYP2C9 and 2C19, as the residue was vertically displaced from the heme in CYP2C9, which would widen the drug binding space. Thus, the E72K mutation in CYP2C19 could induce a 2C9-like orientation of Phe114, which could provide a wider space for drug binding with a water molecule resulting in reverse type I binding.
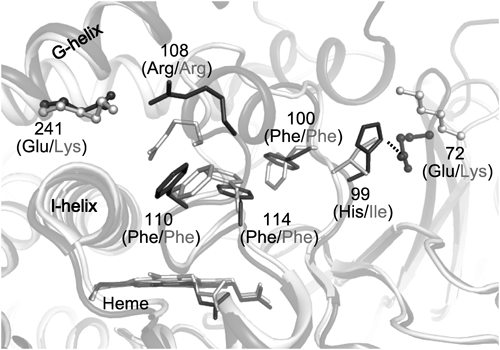
Other mechanisms have been proposed previously.19) Arg47 of CYP102 (P450 BM-3) derived from Bacillus megaterium, which aligns with Lys72 in CYP2C9, was suggested to interact indirectly with acidic substrates with a hydrogen bond network through a water molecule. The hydrogen bond also involved serine 331 (Ser331) corresponding to serine 365 (Ser365) in CYP2C9. Further, the bound substrate might interact with Phe75 by π-stacking. This phenylalanine corresponds to Phe100 in CYP2C9, and that could be the reason why the mutations at residue 72 affected drug binding and metabolism.
Although a hydrogen bond with either His99 or Ser365 was likely to play a key role, the mutations at residue 72 were assumed to cause conformational changes in the B’C-loop resulting in re-orientation of Phe100 and Phe114. The mutations at residue 241 caused slight effects in both CYP2C19 and 2C9. Although the residues 241 were far from the heme, Arg108, which is located in the B’C-loop, was proposed to interact with the residue. Arg108 was also reported to form ionic and hydrogen bonds with aspartic acid 293 (Asp293) and Asn289, respectively.28) Moreover, the Arg=108 residue was shown to be critical for substrate binding.29) This suggests that mutations at position 241 can cause conformational changes in B’C-loop through Arg108, resulting in re-orientation of Phe100 and Phe114 and leading to alterations in drug binding. As a consequence, the mutation at position 241 also had slight effects on drug binding and metabolic activity.
In conclusion, we investigated CYP2C9 and 2C19 WTs and reciprocal mutants on oppositely charged residues. Mutations at positions 72 (CYP2C19 E72K and CYP2C9 K72E mutants) and 241 (CYP2C19 E241K and CYP2C9 K241E mutants) revealed that Glu72 plays a more predominant role than Glu241 in the metabolism of TCA drugs. The effects of mutations were also assessed by plotting the Kd values against the Km values, a kinetics parameter for metabolic activity. The plots were linear, and either the slopes or intercepts could be indicators of the properties of drugs. These results suggest that the reciprocal residue could be critical for drug discrimination in CYP2C19 and 2C9 with the involvement of several residues in the B’C-loop adjacent to the drug binding site.
Acknowledgment
This work was financially supported in part or fully by Grants-in-Aid for Scientific Research (23390011 to TU and 23790045 to TY) from JSPS, Japan. TZA was financially supported from Egyptian government through scholarship.
References
- 1) Attia T. Z., Yamashita T., Miyamoto M., Koizumi A., Yasuhara Y., Node J., Erikawa Y., Komiyama Y., Horii C., Yamada M., Omar M. A., Abdelmageed O. H., Derayea S. M., Uno T., Chem. Pharm. Bull., 60, 1544–1549 (2012).
- 2) Furlanut M., Benetello P., Pharmacol. Res., 22, 15–25 (1990).
- 3) Rudorfer M. V., Potter W. Z., Cell. Mol. Neurobiol., 19, 373–409 (1999).
- 4) Anzenbacher P., Anzenbacherova E., Cell. Mol. Life Sci., 58, 737–747 (2001).
- 5) Hodgson J., Nat. Biotechnol., 19, 722–726 (2001).
- 6) Goldstein J. A., De Morais S. M., Pharmacogenetics, 4, 285–300 (1994).
- 7) Schmidt B., Joussen N., Bode M., Schuphan I., Biochem. Soc. Trans., 34, 1241–1245 (2006).
- 8) Chiba K., Kobayashi K., Manabe K., Tani M., Kamataki T., Ishizaki T., J. Pharmacol. Exp. Ther., 266, 52–59 (1993).
- 9) Goldstein J. A., Faletto M. B., Romkes-Sparks M., Sullivan T., Kitareewan S., Raucy J. L., Lasker J. M., Ghanayem B. I., Biochemistry, 33, 1743–1752 (1994).
- 10) Pearce R. E., Rodrigues A. D., Goldstein J. A., Parkinson A., J. Pharmacol. Exp. Ther., 277, 805–816 (1996).
- 11) Ibeanu G. C., Ghanayem B. I., Linko P., Li L., Pederson L. G., Goldstein J. A., J. Biol. Chem., 271, 12496–12501 (1996).
- 12) Wada Y., Mitsuda M., Ishihara Y., Watanabe M., Iwasaki M., Asahi S., J. Biochem., 144, 323–333 (2008).
- 13) Tsao C. C., Wester M. R., Ghanayem B., Coulter S. J., Chanas B., Johnson E. F., Goldstein J. A., Biochemistry, 40, 1937–1944 (2001).
- 14) Niwa T., Kageyama A., Kishimoto K., Yabusaki Y., Ishibashi F., Katagiri M., Drug Metab. Dispos., 30, 931–936 (2002).
- 15) Jung F., Griffin K. J., Song W., Richardson T. H., Yang M., Johnson E. F., Biochemistry, 37, 16270–16279 (1998).
- 16) Klose T. S., Ibeanu G. C., Ghanayem B. I., Pedersen L. G., Li L., Hall S. D., Goldstein J. A., Arch. Biochem. Biophys., 357, 240–248 (1998).
- 17) Davies C., Witham K., Scott J. R., Pearson A., Devoss J. J., Graham S. E., Gillam E. M., Drug Metab. Dispos., 32, 431–436 (2004).
- 18) Ridderström M., Masimirembwa C., Trump-Kallmeyer S., Ahlefelt M., Otter C., Andersson T. B., Biochem. Biophys. Res. Commun., 270, 983–987 (2000).
- 19) Lewis D. F., Dickins M., Weaver R. J., Eddershaw P. J., Goldfarb P. S., Tarbit M. H., Xenobiotica, 28, 235–268 (1998).
- 20) Marohnic C. C., Panda S. P., Martasek P., Masters B. S., J. Biol. Chem., 281, 35975–35982 (2006).
- 21) Yasukochi Y., Masters B. S., J. Biol. Chem., 251, 5337–5344 (1976).
- 22) Yamashita T., Hoashi Y., Watanabe K., Tomisugi Y., Ishikawa Y., Uno T., J. Biol. Chem., 279, 21394–21400 (2004).
- 23) Uno T., Ryu D., Tsutsumi H., Tomisugi Y., Ishikawa Y., Wilkinson A. J., Sato H., Hayashi T., J. Biol. Chem., 279, 5886–5893 (2003).
- 24) Schenkman J. B., Remmer H., Estabrook R. W., Mol. Pharmacol., 3, 113–123 (1967).
- 25) Shimada T., Kim D., Murayama N., Tanaka K., Takenaka S., Nagy L. D., Folkman L. M., Foroozesh M. K., Komori M., Yamazaki H., Guengerich F. P., Chem. Res. Toxicol., 2013, in press.
- 26) Venkatakrishnan K., Greenblatt D. J., Von Moltke L. L., Schmider J., Harmatz J. S., Shader R. I., J. Clin. Pharmacol., 38, 112–121 (1998).
- 27) Reynald R. L., Sansen S., Stout C. D., Johnson E. F., J. Biol. Chem., 287, 44581–44591 (2012).
- 28) Locuson C. W. 2nd, Wahlstrom J. L., Rock D. A., Rock D. A., Jones J. P., Drug Metab. Dispos., 31, 967–971 (2003).
- 29) Peng C. C., Rushmore T., Crouch G. J., Jones J. P., Bioorg. Med. Chem., 16, 4064–4074 (2008).