Off-target effects, commonly emerged in anticancer drug discovery, often leads to undesired side effects. However, it may also lead to favorable pharmacological profiles in drug re-purposing.1) In kinase-targeted drug discovery, it has been verified that small molecule inhibitors usually bind to multiple targets, including the rarely reported nucleic acid such as double-strand DNA exemplified by flavopiridol.2,3) The off-target effects of kinase inhibitors have always focused on homologous kinases in a dysregulated signaling pathway while rarely on nucleic acid targets at the preliminary stage, although DNA binding affinities, at the late-stage, may be revealed in multi-target small-molecule antibiotic and anticancer drug study. When small molecules bind to the targeted DNA, they alter the normal activity of the DNA with consequent cytotoxicity, cell death, and the abnormal expression of oncogenes.
Intercalation, alkylation and groove binding are three main mechanisms for compounds targeting DNA.4) The intercalation often causes obvious conformational changes of DNA and it occurs non-selectively in different DNA binding sites. For the intercalator, improving molecular planarity would facilitate its embedding to DNA base pairs. Alkylation of DNA is to transfer an alkyl group from exogenous ligand to DNA and form covalent alkyl lesions. So, alkylators can strongly affect the integrity of DNA double strands by forming alkylation cross-linking and preventing uncoiling of the DNA double helices.5) Due to the lack of selectivity, both DNA intercalators and alkylators may cause a potential risk of serious dysfunction of DNA replication, and thus, they are unsuitable as drug candidates.6) Comparatively, the groove binder fit into the minor groove and may form more selective interaction with a specific DNA sequence, as it possesses a larger DNA binding region, such as distamycin, netropsin and Lexitropsins.7,8) Moreover, through non-bonded interactions, groove binders can slide into the narrow and long helical cavity formed by the DNA double strands, less agitating the DNA structure or damaging the integrity of DNA. Therefore, the DNA groove binder represents an important class of anticancer agents.
The pyrazole ring is a useful molecular fragment in drug design, and the pyrazoles have demonstrated diverse pharmacological effects, such as antitumor, analgesic, anti-inflammatory, antipyretic, anti-arrhythmic and antibiosis properties.9) With the aim of increasing the structural diversity of substituted pyrazoles conventional preparation approaches have been developed.10–12) In addition to the creation of C–N and C–C bonds by cross-coupling reactions of aryl electrophiles with substituted pyrazoles, more promising alternative methods have been used in further synthesis and chemical purification with short reaction time and excellent yield, including ultrasound depositing, using condensation reagents and microwave synthesis and so on.13–17) All these greatly promote the progress of research in pyrazole compounds and recently such scaffold is not only employed in developing various kinase inhibitors,10) but also in molecules that directly interact with DNA stands, such as metal complexes, alkylating agents, dimeric analogues, polyamide derivatives and some pyrazole-fused derivatives.18–25) The unexpected interaction between DNA and pyrazole-derived kinase inhibitors can cause off-target effects, including potential improvement of cell toxicity and anticancer activity. Pyrazole ring can form a rigid plane with electron-rich system chromophores and embed into the DNA minor grooves to make a π–π stacking with DNA bases,26–28) which is usually essential for the DNA-binding. Additionally, linking flexible groups to pyrazole-contained scaffold may acquire favorable binding affinities to DNA strands.29,30) As sequence-specific DNA binding ability has been reported in an increasing number of pyrazole analogs,31) more attention should be paid to DNA interaction of pyrazole derivatives in the process of drug development.
In our previous study, we synthesized a series of novel pyrazole derivatives designed as kinase inhibitors to be potential antitumor drugs. Their biological activity evaluations elucidated that these compounds exhibited relatively remarkable cell proliferation inhibition and weak multiple kinase activities.32,33) Hence, besides the inhibition of kinase leading antitumor activity, another mechanism may exist for the compounds’ inhibition of cell proliferation. Meanwhile, enlightened by the potential DNA-binding ability of pyrazole compounds as mentioned above, we hypothesized that the DNA binding of these 1H-pyrazole-3-carboxamide derivatives may contribute to their antitumor activity. To verify our hypothesis and discover possible off-target effects, four most active compounds from the preliminary screen (Chart 1) were synthesized and tested by DNA-binding experiments to reveal the mechanism of their antitumor activity.

Chart 1. Structure of 1H-Pyrazole-3-carboxamide Derivatives
In this paper, we employed a computational method to simulate the binding of these compounds with a known DNA fragment. Considering the energy feasibility on their interactions with DNA predicted by molecular docking, we carried out electronic absorption spectra, fluorescence spectroscopy study, viscosity measurement and DNA cleavage assays to analyze the binding ability and binding mode. Finally, we confirmed that these pyrazole compounds did interact with DNA and the interaction could be an alternative contribution to inhibit cancer cell proliferation by an off-target mechanism.
Experimental
Reagents and MaterialsThe compounds were synthesized according to the following procedure. Ethidium bromide (EB), calf thymus DNA (CT-DNA) and pBR322 plasmid DNA were purchased from Sigma Chemical Co. Tris–HCl/NaCl (5 mM [Tris(hydroxymethyl)aminomethane] and 50 mM NaCl, and adjusted to pH 7.4 with hydrochloric acid) buffer solution was prepared using deionized sonicated triple-distilled water and other chemicals used were of analytical grade. Solution of the compound and other agents used for strand scission were prepared freshly in triple-distilled water before use. Solvents used in this research were purified by standard procedures. The purity and concentration per nucleotide of CT-DNA solution (ε=6600 M−1 cm−1 at 260 nm) in the buffer were checked by a UV-Vis spectrophotometer, and a ratio of UV absorbance of about 1.8–1.9 : 1 at 260 nm and 280 nm indicated that the CT-DNA was sufficiently free of protein.
InstrumentsAll the electronic absorption spectra were recorded on a Shimadzu UV-1800 Visible spectrophotometer after mixing for 5 min at 25°C. And all fluorescence spectra were recorded on a RF-5301PC Spectrofluorimeter (Shimadzu, Japan) equipped with 0.2 cm quartz cells, the widths of both the excitation and emission slit were set to 5 nm at 25°C. The gel was photographed on a capturing system gel printer plus (TDI, Tecnologia para Diagnostico e Investigación, Madrid, Spain). Bands were quantified using the Scion Image for Windows software program based on NHI Kinases and the solution used in assays (cyclin-dependent kinase 2 (CDK2), epidermal growth factor receptor (EGFR) and Abelson tyrosine-protein kinase 2 (ABL2) were purchased from Invitrogen (U.S.A.). KinEASE-TK for Homogeneous Time Resolved Fluorescence (HTRF) was provided by Cisbio (U.S.A.). Reactions were conducted in a 384-well plate (Corning Part #3676).
In Vitro Pharmacological EvaluationAll the compounds were evaluated for their in vitro cytotoxic activity against human hepatocellular liver carcinoma cell Hep2G and human colon cancer cells HCT116 by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay.34) HepG2 and HCT116 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Invitrogen, U.S.A.) supplemented with 10% fetal bovine serum (FBS, Hyclone, U.S.A.), 2 mM L-glutamine, 100 µg/mL streptomycin and 100 U/mL penicillin (Invitrogen). The cells were incubated in a humidified atmosphere with 5% CO2 at 37°C. During logarithmic growth phase, the cells were seeded into 96-well plates to achieve a density of 1×104 cells/well, and cultured for 24 h, followed by exposure to various concentrations of compounds tested for 48 h. Twenty microliter of MTT (Genview, U.S.A.) solution (5 mg/mL) was subsequently added to each well and mixed, then the cells were incubated for an additional 4 h. Culture supernatant was removed and 150 µL of dimethyl sulfoxide (DMSO) (Sangon Biotech, China) was added into each well to make the MTT-formazan crystals completely dissolved. Cell growth inhibition was determined by measuring the absorbance at 570 nm using a microplate reader and calculated according to the equation: Growth inhibition=(1−optical density (OD) of treated cells/OD of control cells)×100%. The half maximal inhibitory concentrations (IC50) were obtained from liner regression analysis for each tested compound, using GraphPad Prism software.
Kinase activity was measured using HTRF assay.35,36) On the Beckman Coulter detection platform, HTRF assays were performed in 384-well plates. The following were combined in the reaction mixture: 4 µL of different concentrations of compound (diluted in 100% DMSO at 50 μM), 8 µL of enzyme, and 8 µL of ATP/substrate mix (final concentrations around Km values). Enzymes, ATP, and substrate were previously diluted in kinase buffer to get final concentrations in the well: 50 mM N-(2-hydroxyethyl)piperazine-N′-2-ethanesulfonic acid (Hepes)/NaOH (pH 7.4), 40 μM Na3VO4, 0.005% ween-20, 1 mM dithiothreitol (DTT), and various concentrations of MnCl2 or MgCl2. The ATP/substrate mixture was added to initiate the assay. The reaction was stopped by addition revelation mixture (55 µL, ethylenediaminetetraacetic acid (EDTA), 0.33 M), specific europium-cryptate antibody antiphospho-ubstrate, and XL-665 with various tags (CisBio International) after being incubated for different times at room temperature. The final concentrations of mixture prepared in revelation buffer in the well: 50 mM Hepes/NaOH (pH 7.4), 0.1% bovine serum albumin (BSA), and 150 mM KF. The reaction was incubated more than 1 h at room temperature, and then detected by Beckman Coulter detection platform. Kinase activities were expressed as a percentage of maximal activity (without compound), resulting from two independent experiments. In all cases, IC50 values were calculated from replicate curves, using GraphPad Prism software. Staurosporine and Roscovitine were used as positive controls in the kinase assay.
Molecular Modeling and DockingMolecular modeling was performed using SYBYL7.3 on a Hewlett-Packard XW4100 PC workstation with a Red Hat Enterprise 4 Linux operating system. All molecules except the DNA were built using SYBYL. The DNA structure was downloaded from the PDB database and prepared by removing both the bound molecules and all waters to avoid potential interference with the docking. Hydrogen was added to the DNA by SYBYL. The compounds were docked into the solution structure of B-DNA dodecamer d(AAT CTT TAA AGA TT)2 (PDB ID:2K4L) with DOCK5.4.37)
Electronic Absorption and Fluorescence SpectraThe DNA-binding experiments were performed at 25°C. Using the electronic absorption spectra, the relative binding of the four complexes to CT-DNA was studied in 5 mM Tris–HCl/50 mM NaCl buffer (pH=7.4). The compounds were dissolved in a solvent of Tris–HCl buffer (5 mM Tris–HCl, 50 mM NaCl, pH 7.4) at the concentration 1.0×10−5 M. Absorption titration experiments were performed with fixed concentration drugs (10 mM) while gradually increasing the concentration of CT-DNA. The ratio of CDNA/Ccompound in each curve decreased as follows: 6/100, 13/100, 25/100, 38/100, 51/100, 76/100, 102/100, 127/100, 152/100, 230/100, 254/100. In order to eliminate the absorbance of CT-DNA itself, an equal amount of CT-DNA was added to both the compound solution and the reference solution during measuring the absorption spectra. By the fluorescence spectral method, the relative binding of compounds to CT-DNA were studied with an EB-bound CT-DNA solution in 5 mM Tris–HCl/50 mM NaCl buffer (pH=7.4). The ratio of CEB-CT-DNA/Ccompound in each curve decreased as follows: 0, 1/10, 1/5, 3/10, 2/5, 1/2, 3/5, 4/5 and 1/1 (Fig. 6). The excitation wavelength was 520 nm for all cases.
Viscosity ExperimentsViscosity measurements were carried on an Ostwald’s viscometer at 30±0.1°C in a constant temperature bath. The DNA concentration was fixed at 1.5×10−4 M and the flow time were measured with a digital stopwatch; the mean values of three measurements were used to evaluate the viscosity of each sample. Data are presented as (η/η0)1/3 versus r (r=[compound]/[DNA]=0–1), where η is the viscosity of DNA in the presence of compound and η0 is the viscosity of DNA alone. Viscosity values were calculated from the observed flowing time of DNA-containing solutions (t) corrected for that of the buffer alone (t0), η=(t−t0)/t0.
pBR322 DNA Cleavage AssaysThe plasmid DNA cleavage experiments were performed using pBR322 DNA in Tris–HCl buffer. Reactions were performed by incubating DNA (0.05 mM bp) at 37°C in the presence or absence of the compound for the indicated time periods. All reactions were quenched by loading buffer. Agarose gel electrophoresis was carried out on a 1% agarose gel in 0.5× TAE (Tris–acetate–EDTA) buffer containing 0.5 µg/mL EB at 80 V for 1.5 h, then the gel image was captured by the Scion Image System.
Results and Discussion
SynthesisThe synthetic pathways adopted for the preparation of title compounds are illustrated in Chart 2. pym-5, pym-55 and pym-n all bearing a characteristic functional group have a common intermediate (5), which was prepared through alkylation, reduction and acylation. The reduction step of the nitro group to amide employed hydrazine hydrate in the presence of an iron(III) oxide hydroxide (FeO(OH)/C) as a catalyst38) in the refluxing 95% ethanol to get a high yield product within 2 h.39–41) Furthermore, the catalyst can be recycled after the reaction through washing with anhydrous ethanol. pym-5 bearing a urea group was synthesized from compound 5 with cyclopropylamine employed CDI. pym-55 was prepared using N-hydroxybenzotriazole (HOBT) and 1-ethyl-(3-(3-dimethylamino)propyl)carbodiimide hydrochloride (EDCI) as condensing agent to acylate compound 5 by benzonic acid. pym-n was gained through catalyzed hydrogenation with NaBH4 from the intermediate of Schiff base which was generated by mixing compound 5 with pyridine formaldehyde. The preparation of pyz-5 was similar to pym-5 with the starting reactant morphine instead of N-methylpiperazine.

4-Amino-N-[4-[(4-methylpiperazin-1-yl)methyl]phenyl]-1H-pyrazole-3-carboxamide (5): The mixture of N-methyl piperazine (4.9 mL, 44.2 mmol) and triethylamine (12 mL, 86.3 mmol) in dichloromethane (20 mL) was added dropwise to a stirred solution of Nitro-benzyl bromide 1 (10.0 mg, 46.3 mmol) in dichloromethane (100 mL) under the ice-water bath and was refluxed for 1 h. The reaction mixture was extracted with chloroform (100 mL×3), washed with water and saturated sodium chloride each time (100 mL×1). The organic phase was dried with anhydrous magnesium sulfate, filtered, evaporated under vacuum to give pale yellow solid 2. Without further purification, 2 mixed with FeO(OH)/C (catalyst 2.0 g) and 95% ethanol (100 mL) were kept refluxing and dropwise added the mixture of hydrazine hydrate (25 mL) and 95% ethanol (20 mL), then filtrated when the solution was hot. The residue was washed with hot ethanol (30 mL×2). The solvent was distilled under vacuum to give white solid 3 (6.7 g), then mixed with 4-nitro-1H-pyrazole-3-acid (6.3 g, 40.0 mmol), EDCI (8.4 g, 43.8 mmol) and HOBT (6.0 g, 44.4 mmol) in anhydrous DMF (100 mL), stirred for 24 h at room temperature. The reaction mixture was poured into ice water (200 mL). A large amount of yellow solid precipitation was acquired. The pure product 4 was got from recrystallizing with mixed solvent of methanol and ethyl acetate, then the same process of hydrazine hydrate reduction was done with the catalyst of FeO(OH)/C. The solvent was distilled under vacuum to give white solid 5 (3.5 g). Yield=63.9%; mp 199–201°C; IR (KBr) cm−1: 3369, 3227 (NH2), 1346 (C=C), 1456 (C=N) pyrazole, 646 (ArH); 1H-NMR (300 MHz, DMSO-d6) δ (ppm): 2.1 (3H, s, –CH3), 2.3–2.5 (8H, m, –CH2–), 3.3 (2H, s, –CH2–), 4.7 (1H, s, –NH2), 7.1–7.2 (3H, m, ArH), 7.7 (2H, d, J=10.5 Hz, ArH), 9.7 (1H, s, –NHCO–), 12.7 (1H, s, Pyrazole); MS m/z: 315.82 (M+); Anal. Calcd for C16H22N6O: C, 61.13; H, 7.05; N, 26.73; O, 5.09. Found: C, 61.31; H, 6.84; N, 26.52.
5-(3-Cyclopropylureido)-N-[4-[(4-methylpiperazinyl)methyl]phenyl]-1H-pyrazole-3-carboxamide (pym-5): A mixture of 5 (314.0 mg, 1.0 mmol), CDI (210.6 mg, 1.3 mmol) in anhydrous DMF (20 mL) was kept refluxing. Cyclopropylamine (0.5 mL) was added to the reaction solution after cooling down to room temperature. The solution was stirred at room temperature for 3 h, evaporated under vacuum to give pale yellow oily substance. The crude product was separated by column chromatography (developing solvent: methanol–chloroform 1 : 30) to get pym-5 (222.0 mg). Yield=56.1%; mp 156–158°C; IR (KBr) cm−1: 2995 (CH2) cycloproyl, 1656 (C=O), 1280 (C–N) uera, 3371 (NH), 1344 (C=C), 1454 (C=N) pyrazole; 1H-NMR (300 MHz, DMSO-d6) δ: 0.4–0.6 (4H, m, –CH2–), 2.1 (3H, s, –CH3), 2.3–2.7 (9H, m, –CH2–, –CHN–), 3.3 (2H, s, –CH2–), 7.2 (2H, d, J=8.4 Hz, ArH), 7.3 (1H, s, ArH), 7.7 (2H, d, J=8.4 Hz, ArH), 8.0 (1H, s, –NHCONH–), 8.78 (1H, s, –NHCONH–), 10.0 (1H, s, –NHCO–), 13.1 (1H, s, pyrazole); MS m/z: 396.94 (M−); Anal. Calcd for C20H27N7O2: C, 60.44; H, 6.85; N, 24.67; O, 8.05. Found: C, 60.32; H, 7.06; N, 24.45.
N-[4-[(4-Methylpiperazin-1-yl)methyl]phenyl]-5-(phenylamino)-1H-pyrazole-3-carboxamide (pym-55): Compound 5 (314.0 mg, 1.0 mmol), benzoic acid (112.0 mg, 1.3 mmol), EDCI (249.2 mg, 1.3 mmol), HOBT (175.7 mg, 1.3 mmol) were mixed in anhydrous DMF (30 mL), stirred 24 h at room temperature. The solvent was evaporated under vacuum to give pale oily substance, the crude product was separated by column chromatography (developing solvent: methanol–chloroform 1 : 50) to get pym-55 (200.3 mg). Yield=48.7%; mp 236–238°C; IR (KBr) cm−1: 3369 (–NH–) Amie, 3335 (NH), 1446 (C=N), 1377 (C=C) pyrazole, 1593 (ArH), 1656 (O=C–NH), 1174 (C–N); 1H-NMR (300 MHz, DMSO-d6) δ: 2.1 (3H, s, –CH3), 2.4–2.6 (8H, m, –NCH2–), 4.1 (2H, s, –CH2–), 6.9–7.0 (3H, m, ArH), 7.1 (2H, d, J=8.7 Hz, ArH), 7.2 (1H, m, ArH), 7.7 (2H, d, J=8.7 Hz, ArH), 8.0 (1H, s, ArH), 8.2 (1H, m, ArH), 8.9 (1H, s, –NHCO–), 10.0 (1H, s, –NHCO–), 13.2 (1H, s, pyrazole). MS m/z: 419.30 (M+); Anal. Calcd for C23H26N6O2: C, 66.01; H, 6.26; N, 20.08; O, 7.65. Found: C, 65.98; H, 6.43; N, 20.35.
N-[4-[(4-Methylpiperazin-1-yl)methyl]phenyl]-5-(pyridin-4-yl-methyl)-1H-pyrazole-3-carboxamide (pym-n): The mixture of 5 (314.0 mg, 1.0 mmol), pyridine formaldehyde (0.5 mL) and methanol (30 mL) was heated to reflux, then cooled to room temperature. An excess sodium borohydride (500.0 mg) was added, then the reaction mixture was kept refluxing for 2 h andevaporated under vacuum to give pale yellow oily substance and separated by column chromatograph (developing solvent: methanol–chloroform 1 : 20). Yield=48.6%; mp 262–264°C; IR (KBr) cm−1: 3369 (–NH–), 3335 (NH), 1456 (C=N), 1348 (C=C), pyrazole, 1593 (ArH), 1666 (O=C–NH), 1130 (C–N). 1H-NMR (300 MHz, DMSO-d6) δ: 2.2 (3H, s, –CH3), 2.3–2.5 (8H, m, –CH2–), 3.4 (2H, s, –CH2–), 4.4 (2H, s, –CH2–), 5.6 (1H, m, –NH–), 6.9 (2H, m, ArH), 7.2 (2H, d, J=8.4 Hz, ArH), 7.3 (1H, s, ArH), 7.4 (2H, d, J=8.7 Hz, ArH), 7.7 (2H, d, J=8.4 Hz, ArH), 9.7 (1H, s, –NHCO–), 12.8 (1H, s, pyrazole); MS m/z: 406 (M+); Anal. Calcd for C22H27N7O: C, 65.16; H, 6.71; N, 24.1; O, 3.95. Found: C, 65.32; H, 6.90; N, 24.25.
5-(3-Cyclopropylureido)-N-[4-(morpholinomethyl)phenyl]-1H-pyrazole-3-carboxamide (pyz-5): The preparation process of pyz-5 which began with morphine was the same as pym-5. Yield=56.1%; mp 121–123°C; IR (KBr) cm−1: 3335 (NH), 1344 (C=C), 1454 (C=N) pyrazole, 2995 (CH2) cycloproyl, 1666 (O=C–NH), 1533, 1265 (amide), 1066 (C–O) ether; 1H-NMR (300 MHz, DMSO-d6) δ: 0.7–1.0 (4H, m, –CH2–), 2.5 (4H, m, –NCH2–), 2.7 (1H, m, –CH–), 3.3 (2H, s, –CH2–), 3.6 (4H, m, –OCH2–), 7.0 (1H, m, ArH), 7.2 (2H, d, J=8.7 Hz, ArH), 7.7 (2H, d, J=8.7 Hz, ArH), 10.0 (1H, s, –NHCO–), 11.3 (1H, s, –NHCONH–), 12.0 (1H, s, –NHCONH–), 14.0 (1H, s, pyrazole); MS m/z: 383.90 (M−); Anal. Calcd for C19H24N6O3: C, 59.36; H, 6.29; N, 21.86; O, 12.49%. Found: C, 59.65; H, 6.63; N, 21.55.
In Vitro Pharmacological EvaluationThe antiproliferative activity of the four selected compounds was evaluated by MTT tetrazolium dye assays on HCT116 cells and HepG2 cells (Table 1). Among the four compounds, the inhibition IC50 values of pym-5 and pym-55 are similar to the positive drug Iressa (IC50=9.34 µM, HCT116) and Roscovtine (IC50=9.87 µM, HepG2) respectively. However, the targeted kinase inhibition IC50 values for CDK2, EGFR and ABL2 of four compounds were not comparable to the positive drug.
Table 1. Antiproliferative Activity and Kinase Inhibition of Four Derivatives
| Compd. | Cell line inhibition IC50a) (µM) | Kinase inhibition IC50a) (µM) |
|---|
| HepG2 | HCT116 | CDK2 | EGFR | ABL2 |
|---|
| pym-5 | 32.23 | 15.94 | 7.23 | 3.31 | 2.05 |
| pym-n | 89.5 | 10.93 | 9.5 | 13.51 | 3.98 |
| pym-55 | 15.96 | NT | 10.96 | 32.67 | 12.24 |
| pyz-5 | 244.18 | NT | 6.18 | 7.92 | 2.34 |
| Iressa | NT | 9.34 | NT | NT | NT |
| Roscovitine | 9.87 | NT | 0.01 | NT | NT |
| Staurosporine | NT | NT | NT | 0.11 | 0.054 |
a) Values are the mean from two independent dose–response curves; variation was generally (±25%) with the reference compounds with Iressa and Roscovitine. NT, not tested.
Molecular docking was applied to investigate the molecular mechanism of DNA-ligand binding. The four compounds all interact with the base pairs of DNA via groove binding mode. Docking modes of the four compounds are similar, and the detailed view of the DNA binding mode of pym-5 is shown in Fig. 1. The molecule is shaped as a chain by two amide bonds on the pyrazole ring, which enables the molecule to properly stretch along the DNA minor groove. Furthermore, acetyl groups are conjugated with the whole aromatic heterocycle, leading this part of the molecule to be a planar rigid system which could exactly extend into the space shaped by the adjacent base pairs and benefit the binding through van der Waals. The benzene ring stacks with two adenine bases in a parallel displaced conformation for π stacking interactions.

Fig. 1. The Docking Mode of Compound pym-5 with DNA in the Minor Groove
The binding energy values for pym-5, pyz-5, pym-55 and pym-n were scored as −49.67, −44.56, −30.91, −28.80 kcal/mol, respectively. Among them, pym-5 had the strongest binding affinity with the lowest binding energy. Compared with pym-n and pym-55, the cyclopropyl group of pym-5 formed a smaller steric-negative interaction with DNA, so that pym-5 fit the minor groove of DNA more snugly and exhibited a higher binding affinity. Also, at physiological pH, the protonation of N atom on the piperazine ring is more favorable than the morpholine of pyz-5 to interact with the electronegative phosphorous group. In addition, the pyrazole plane of pym-5 formed a weak σ–π stacking with the C4′ of sugar residues compared to others, and meanwhile, the potential hydrogen bonds could be formed between urea group and DNA. Both interactions may influence the conformation of the phosphate-deoxyribose backbone. Due to the factors stated above, pym-5 would penetrate deeply into the minor groove of DNA and exhibit a better DNA binding affinity.
Electronic Absorption Spectral StudiesThe potential binding ability of four pyrazole compounds (pym-5, pyz-5, pym-55 and pym-n) to calf thymus DNA (CT-DNA) was studied by electronic absorption spectroscopy. The UV-Vis spectra were measured in the way: Fixing the concentration of compounds (Ccompound=3.00×10−5 M) while increasing the concentration of CT-DNA. The tendency of hypochromism without obvious red shift occurred in four compounds can be observed in Fig. 2, which suggests that the interaction of compounds to DNA is not from a classic intercalation mechanism but probably groove binding mode.42,43) The absorption relationship between the compounds and DNA can be expressed by double reciprocal equation44):
 | (1) |
 | (2) |
 | (3) |
Where A0 and A are the absorbance of compounds without and in the presences of DNA, respectively. Kb is the binding constant between the compounds and DNA, and CDNA is the concentration of DNA. In the plot of ∆A versus 1/CDNA (Fig. 3), Kb is then given by the ratio of the intercept to slope intercept listed in Table 2, which indicates that the docking result of the DNA binding order is in accordance with the result evaluated by electronic spectra that is pym-5>pyz-5>pym-55>pym-n.
Table 2. The Docking Results and DNA-Binding Constants
| Compound | pym-5 | pyz-58 | pym-55 | pym-n |
|---|
| Binding energy (kcal/mol) | −49.67 | −44.56 | −30.91 | −28.80 |
| Binding constant K (L/mol) | 1.06×105 | 8.83×104 | 6.85×104 | 1.64×104 |
With higher sensitivity and accuracy, fluorescence spectroscopic study was carried out to further clarify the interaction mode of four compounds with DNA. No luminescence was observed at room temperature in the aqueous solution, neither in any organic solvent examined. Thus, the experiment was carried out in the EB background. EB molecule can make a strong interaction with the adjacent DNA base pairs and emit intense fluorescence at about 550–650 nm in the hydrophobic environment provided by the groove of DNA. This fluorescence would be quenched when the compound squeezed out of the EB molecule from the hydrophobic region and exposed the EB molecule to the polarity micro-environment by inserting into the groove or the internal structure of the DNA. Therefore, the quenching degree can be used to measure the binding propensity of the compound to DNA. As the concentration of the compound was increased, all the compounds showed distinctive decrease happened to the intensity of EB–CT-DNA system. The most interesting spectrum was pym-5, in Fig. 4, more than 50% decrease of the emission intensity of the EB–CT-DNA (1×10−4 M) complex with the compound pym-5 (3×10−5 M) being added, which indicates a strong interaction between pym-5 and DNA. Based on the classical theory of fluorescence quenching: Sterm–Volmer equation45)

Fig. 4. Representative Fluorescence Emission Spectra of DNA-EB in Tris–HCl Buffer (pH 7.4) with the Increasing in Molar Ratio of pym-5 to DNA (0, 1/10, 1/5, 3/10, 2/5, 1/2, 3/5, 4/5, 1/1) at 25°C
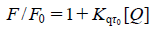 | (4) |
The plot of F0/F versus [Q] ([Q] is the concentration of quencher compound) is linear for either simple dynamic quenching or static quenching. Hence, a broken line shown in Fig. 5a reveals that two dominant quenching processes exist in the system possibly.46) The turning point was found at the concentration of 4×10−5 M. That means the mechanism of the quenching process changed at this concentration. Kq (1.8×1012 M −1/s) given by the ratio of the slope to intercept in the second stage of this plot is greater than other kinds of quencher (the maximum diffusion of biological macromolecules sudden collision off constant Kq is 2×1010 M−1/s), suggesting that the second quenching is a static quenching process.47) The binding constant K and the site number of n were calculated by Eq. (5),48) K and n were 8.57×105 M−1 and 1.38, respectively shown in Fig. 5b.

Fig. 5. (a) Stern–Volmer Plots for the Fluorescence Quenching of the EB–CT-DNA Complex with the Compound pym-5 and (b) log((F0−F)/F) of log[Q] for the Linear
 | (5) |
Spectra probes generally provide necessary, but not more persuasive evidence than viscosity measurement. The value of viscosity is sensitive to the length change of DNA. The distance between adjacent base pair will be larger, when the molecule intercalates into DNA, causing obviously lengthening. In other way of interaction model (electrostatic attraction or groove binding), due to no increase in the DNA length the viscosity of the sample solution changes litter. In Fig. 6, the minor change in viscosity for compounds is an indication of groove binding.

Fig. 6. Effect of Increasing Amounts of Compound on the Viscosity of CT-DNA (1.5×10−4 M) in Tris–HCl Buffer (r=0.0, 0.05, 0.10, 0.25, 0.3, 0.45, 0.6, 0.8, 1)
Based on the strong ability to bind to the CT-DNA, the four compounds were employed to incubate with pBR322 DNA under the reaction conditions to assess the chemical nuclear activity. The cleavage reaction can be monitored by agarose gel electrophoresis. When circular pBR322 DNA is subjected to electrophoresis, relatively fast migration will be observed in the intact supercoiled form (Form I). Scission of one strand (nicking) makes the Form I relax to generate a slower-moving nicked form (Form II) that we can find in Fig. 7 (pBR322 DNA hydrolytic cleavage promoted by pym-5). Meanwhile, other three compounds performed no cleavage effect in the assay. The conversion of Form I to Form II is observed in a concentration-dependent manner for pym-5 in Fig. 7a.49) A control sample was run in lane 1 containing only buffered DNA at the same concentration of the digested assays. The Form II appeared when the concentration of pym-5 was added to 5×10−7 M. This cleavage activity of pym-5 may be attributed to a hydrolysis of pBR322DNA based on the nucleophile attacking phosphonates by the uramido group. Also, cationic protonated tertiary amine under the pH 7.4 may stabilize the negatively charged nuclear phosphorus penta-coordinated transition state.50)

Fig. 7. Agarose Gel Electrophoresis Patterns for the Cleavage of pBR 322 Plasmid DNA (0.25 µg/µL) with the Addition of Four Compounds Separately in Different Concentration under Dark for 1.5 h, the Result Shown in a (pym-5), b (pym-55), c (pym-n), d (pyz-5) and Lane 1: DNA Control; Lane 2: (1×10−11 M); Lane 3: (5 ×10−10 M); Lane 4: (5×10−9 M); Lane 5: (5×10−8 M); Lane 6: pym-5 (5×10−7 M); Lane 7: (5×10−6 M); Lane 8: (5×10−5 M); Lane 9: (5×10−4 M); Lane 10: (5×10−3 M)


