Abstract
The synthesis of a small series of 2-nitroimidazoles in which the β-amino alcohol side chain was amidated with a range of alkylating/acylating functionality is described. Synthetic methodologies were developed that generally provided for selective N-acyl versus N,O-bisacyl products. In vitro, target analogs showed minimal radiosensitization activity, with only a few exhibiting a sensitizer enhancement ratio (SER) >2.0 and C1.6 values comparable to reference agents RB-6145 and RSU-1069. In an assay to determine potential to alkylate biomolecules, representative analogs showed <1% of the alkylating activity of RSU-1069. In vivo, one analog showed an enhancement ratio of 1.6 relative to vehicle control when tested in B6C3F1 mice with an implanted KHT sarcoma. The data reinforce prior findings that there is a correlation between alkylation potential and in vivo activity.
Several years ago, the dual function 2-nitroimidazole RB-6145 (1a, Fig. 1) was reported to demonstrate excellent radiosensitizing activity in preclinical models of tumor hypoxia.1) Compound 1a is a prodrug of RSU-1069 (2), which has an aziridine moiety in the side chain and was shown to have a high differential toxicity toward hypoxic cells compared to oxic cells under in vitro and in vivo experimental conditions. Additional studies showed that 1a was considerably less emetic than 2 (which had been briefly evaluated in a Phase I clinical trial2)) and thus was chosen for further development as its R-enantiomer (1b, CI-1010). Toward this end, we reported on a scaleable synthesis of 1b.3) Selective, bioreductive metabolism of CI-1010 in hypoxic cells was shown to increase DNA alkylation, and form cross links and strand breaks.4) Despite its efficacy in killing tumors, CI-1010 produced a selective and irreversible degeneration of the outer nuclear and photoreceptor layers of the retina in relevant preclinical animal models. Mechanistic studies showed that CI-1010 induced cell death in photoreceptors and provided evidence in support of a p53-linked activation of caspase-3 in response to DNA damage caused by CI-1010.5) This toxicity was manifested at concentrations required for therapeutic efficacy, thus leading to the termination of the preclinical development of CI-1010.

Fig. 1. Structures of Preclinical Dual Function 2-Nitroimidazole Radiation Sensitizers
The high reactivity of the aziridinyl moiety of 2 toward biological nucleophiles led us to consider the installation of other alkylation/acylation functionality off the 2-nitroimidazole core, which might be manifested in lower toxicity. In this paper, we report on the synthesis of a small series of 2-nitroimidazoles in which the amino alcohol side chain is amidated with a range of head pieces (Michael acceptors, chloromethyl amide, β-lactam) representing a spectrum of predicted alkylating/acylating reactivity, based on the concept of “soft” drug design developed by Bodor.6)
Results and Discussion
ChemistryThe synthetic methodologies utilized to make our 2-nitroimidazole analogs are shown in Fig. 2. The key intermediate in our synthesis was the 2-nitroimidazole β-amino alcohol 4, which was prepared from epoxide 3 in a 62% yield over two steps. We repeated this preparation several times on a modest scale with reproducible results. A related process starting from 2-nitroimidazole has been described relatively recently on a smaller scale.7) The amidation of 4 was then examined under a range of conditions. The goal was to find conditions that would provide selective N- versus O-acylation. For the synthesis of 5a and b, we first examined literature conditions that utilize acid chlorides for the selective acylation of β-amino alcohols.8,9) These surprisingly gave products in extremely poor yield (<10%), which we attribute to additional sites of reactivity from the weakly nucleophilic nitrogens of the nitroimidazole ring. Thus we directed our efforts toward finding milder, less reactive acylating conditions for our couplings. Those shown in Fig. 2 (Methods A–C) provided desired compounds, which could be easily purified by silica gel flash chromatography, in poor to good yields (13–64% following crystallization) and generally with high or complete N-selectivity. Thus, target compounds 5a, b were best accessed via acylation of 4 with anhydrides (Method A) using literature conditions to selectively acylate β-amino alcohols.10) For 5b we observed selective N-acylation, whereas for 5a TLC showed formation of 20–25% N,O-bisacylated product, which could easily be converted in situ to 5a with potassium carbonate in refluxing methanol. Since anhydrides of most of our other acyl side chains were not commercially available, we decided to develop coupling conditions that would allow us to utilize carboxylic acids. Prior reports of bis(2-oxo-3-oxazolidinyl)phosphinic chloride (BOP chloride)11) and diethyl cyanophosphonate12) as mild condensing agents prompted us to try these, leading to 5c–i (Method B) and 5j–l (Method C), respectively. Methods to selectively acylate β-amino alcohols with acrylic acids using BOP chloride have never been described, whereas the use of diethylcyanophosphonate for this purpose appeared recently in the patent literature by Japanese workers.13) Reaction with BOP chloride provided complete N-selectivity for all examples except 5d and e, which gave 10–15% of N,O-bisacylated product. By contrast, reactions with diethyl cyanophosphonate showed complete N-selectivity, perhaps because Method C is the only one in which a polar solvent (N,N-dimethylformamide (DMF)) is used. Thus, we prefer Method C because of its N-selectivity, ease of work-up and known lack of racemization for chiral side chains (e.g., 5i). No attempts were made to optimize reaction yields since our main focus was to synthesize enough of each target compound in high purity for testing. The condensing agents used for Methods B and C complement other reagents (1-ethyl-3-(3-dimethylaminopropyl)carbodiimide, diisopropylcarbodiimide) that are frequently used to acylate β-amino alcohols with acrylic acids.14,15)
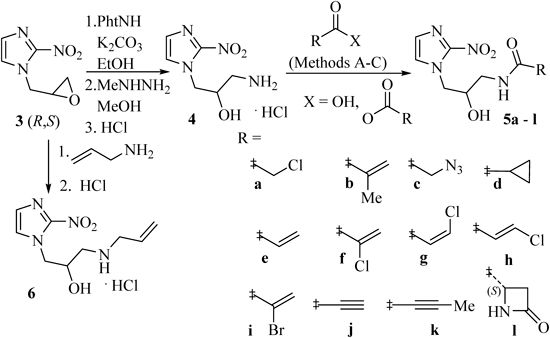
All precursor anhydrides and acids were commercially available except for (S)-4-oxo-2-azetidinecarboxylic acid, which was synthesized by the procedure of Baldwin et al.16) We also aminated epoxide 3 with allylamine to give 6 in 67% yield. We view the terminal olefin as a convenient functional group to introduce electrophilic functionality onto the amine side chain toward making homologous analogs of 1a.
The structures of all target compounds were rigorously established through 1H-NMR, IR, and mass spectrometry, and through combustion analysis.
Biological EvaluationTable 1 contains data for in vitro radiosensitization and relative alkylation capability for the compounds of this study. Since sensitizer enhancement ratios (SER) were determined at the maximum nontoxic dose of each compound, the cell killing observed reflects only the radiosensitizing capability of the compounds. Most target compounds showed minimal radiosensitizing activity. Only 4 of 13 compounds evaluated (5a, g, h, 6) exhibited a sensitizer enhancement ratio >2.0. Compounds which yielded an SER ≥1.8 (5a, b, f, h, 6) were further evaluated in a dose–response study (C1.6 determination). The C1.6 of a compound is a measure of its efficiency of radiosensitization and is defined as the concentration required to give an SER of 1.6. Despite being generally weak as radiosensitizers, three compounds (5f, h, 6) showed C1.6 values that are comparable to or less than reference agents 1a and 2, signifying significant efficiency.
Table 1.
In Vitro Radiosensitizing Activity
a) and Alkylation Potential of 2-Nitroimidazoles
b)| Compound | Max. tolerated conc. (mM) | SER | C1.6 (mM) | Relative alkylation |
|---|
| 1a | 3.0 | 2.8 | 0.5 | NDc) |
| 2 | 0.8 | 2.0 | 0.33 | 100 |
| 5a | 3.0 | 2.4 | 0.71 | <1 |
| 5b | 3.0 | 1.8 | 2.1 | <1 |
| 5c | 3.0 | 1.5 | ND | ND |
| 5d | 1.5 | 1.2 | ND | <1 |
| 5e | 3.0 | 1.3 | ND | <1 |
| 5f | 0.09 | 1.8 | 0.08 | <1 |
| 5g | 3.0 | 2.4 | 0.98 | <1 |
| 5h | 1.5 | 2.2 | 0.38 | <1 |
| 5i | 0.02 | 1.2 | ND | ND |
| 5j | 3.0 | 1.6 | ND | ND |
| 5k | 3.0 | 1.4 | ND | ND |
| 5l | 3.0 | 1.0 | ND | ND |
| 6 | 3.0 | 2.5 | 0.31 | <1 |
a) Values represent the mean of replicate experiments agreeing within ±20%. b) See Supplemental Information for in vitro methods. c) ND=not determined.
We then utilized an established assay17) to determine the relative alkylating activity of representative compounds (5a, b, d–h, 6) versus RSU-1069 (2). Each of these showed <1% of the alkylating activity of RSU-1069. Due to their generally poor radiosensitizing effects and lowered propensity to alkylation, we did not carry out studies to determine if any of the analogs showed selective cytotoxicity under hypoxic conditions.
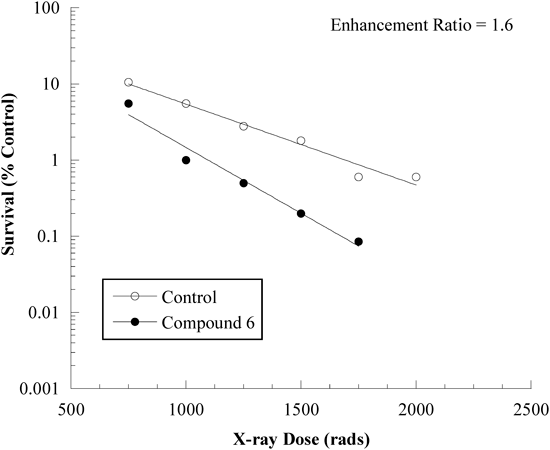
Since compound 6 showed an overall in vitro profile (SER, C1.6) similar to reference agent 1a (RB-6145), we subjected it to in vivo evaluation in B6C3F1 mice with an implanted KHT sarcoma. At an optimal dosing interval prior to irradiation, 6 showed a modest enhancement ratio of 1.6 relative to vehicle control (Fig. 3). In comparison, 1a and 2 (RSU-1069) give ratios of 2.4 and 2.2, respectively, at their maximum tolerated doses. Compounds 5a and g were also tested against this tumor model, but demonstrated little to no activity. Therefore, despite the promising SER activity of these compounds determined in vitro (Table 1), their lack of alkylating potential correlates with diminished in vivo activity.
Conclusion
We have synthesized a small series of 2-nitroimidazoles in which the β-amino alcohol side chain was amidated with a range of head pieces representing a spectrum of predicted alkylating/acylating reactivity. Synthetic methodologies were developed that provided for either selective (Methods A and B) or complete (Method C) N-acylation versus N,O-bisacyl side products. Most target compounds showed minimal radiosensitizing activity and each showed <1% of the alkylating activity of the clinical agent RSU-1069 (2). Further, there is no discernible structure–activity relationships (SAR) for this relatively small series of compounds. One general pattern is evident, however. The marked reduction of alkylation potential relative to RSU-1069, taken together with the general lack of significant activity in standard assays for radiosensitizers, reinforce prior findings that there is a correlation between alkylation potential and in vivo activity.18)
Experimental
General chemistry methods, synthesis procedures, spectral data, and biological methods are given in Supplemental Information.
References
- 1) Jenkins T. C., Naylor M. A., O’Neill P., Threadgill M. D., Cole S., Stratford I. J., Adams G. E., Fielden E. M., Suto M. J., Stier M. A., J. Med. Chem., 33, 2603–2610 (1990).
- 2) Bremner J. C. M., Cancer Metastasis Rev., 12, 177–193 (1993).
- 3) Sercel A. D., Beylin V. G., Marlatt M. E., Leja B., Showalter H. D. H., Michel A., J. Heterocycl. Chem., 43, 1597–1604 (2006).
- 4) Jenner T. J., Sapora O., O’Neill P., Fielden E. M., Biochem. Pharmacol., 37, 3837–3842 (1988).
- 5) Miller T. J., Schneider R. J., Miller J. A., Martin B. P., Al-Ubaidi M. R., Agarwal N., Dethloff L. A., Philbert M. A., Neurotoxicology, 27, 44–59 (2006).
- 6) Bodor N., Adv. Drug Res., 13, 255–331 (1984).
- 7) Lee D. Y.-W., Ji X.-S., Raleigh J. A., U.S. Patent 20080102026A1 (2008).
- 8) McKnight S. L., Pieper A. A., Ready J. M., De Brabander J. K., U.S. Patent 20130040977A1 (2013).
- 9) Glennon R. A., Bondarev M. L., Khorana N., Young R., May J. A., Hellberg M. R., McLaughlin M. A., Sharif N. A., J. Med. Chem., 47, 6034–6041 (2004).
- 10) Myers A. G., PCT Int. Appl. WO9525714A1 (1995).
- 11) Colucci W. J., Tung R. D., Petri J. A., Rich D. H., J. Org. Chem., 55, 2895–2903 (1990).
- 12) Takuma S., Hamada Y., Shioiri T., Chem. Pharm. Bull., 30, 3147–3153 (1982).
- 13) Igarashi Y., Japanese Patent Appl. 2012/116786 A (2012).
- 14) Park J.-J., Lee J. H., Li Q., Diaz K., Chang Y.-T., Chung S.-K., Bioorg. Chem., 36, 220–228 (2008).
- 15) Kummer D. A., Chain W. J., Morales M. R., Quiroga O., Myers A. G., J. Am. Chem. Soc., 130, 13231–13233 (2008).
- 16) Baldwin J. E., Adlington R. M., Russell A. T., Smith M. L., Tetrahedron, 51, 4733–4762 (1995).
- 17) Bodor N., Kaminski J. J., Worley S. D., Gerson S. H., Z. Naturforsch., B: Anorg. Chem. Org. Chem., 35B, 758–763 (1980).
- 18) Suto M. J., Stier M. A., Werbel L. M., Arundel-Suto C. M., Leopold W. R., Elliott W. E., Sebolt-Leopold J. S., J. Med. Chem., 34, 2484–2488 (1991).