Notes
Aliphatic Substitution of o-Carboranyl Phenols Enhances Estrogen Receptor Beta Selectivity
2014 年 62 巻 4 号 p. 386-391
詳細
2014 年 62 巻 4 号 p. 386-391
The two subtypes of estrogen receptor (ER), ERα and ERβ, differ greatly in expression pattern and biological functions, and ERβ-selective ligands candidates to treat immune-related disorders. ERβ-selective ligands have mostly been designed based the idea of introducing a substituent that interferes sterically with the ligand’s interaction with Met421 to selectively decrease the affinity for ERα (the equivalent residue in ERβ is Ile373). Therefore, we designed and synthesized a series of carboranyl phenol derivatives bearing an aliphatic substituent as candidate ERβ-selective ligands. Introduction of a longer aliphatic substituent into the carboranyl moiety enhanced the ERβ selectivity of o-carboranyl phenol derivatives 4, but not m-carboranyl bisphenol derivatives 5. Compound 4c showed 7.4-fold ERβ selectivity in ER-binding assay and exhibited moderate estrogenic activity in cell proliferation assay using MCF-7 cell line.
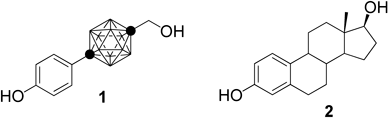
We have shown that the carborane cage is available as a novel hydrophobic pharmacophore for drug development,1) and since then, various carborane-containing bioactive compounds have been reported.2,3) Prior to that, carboranes had been used only as boron carriers for boron neutron capture therapy (BNCT), based on their high boron content.4) We also designed and synthesized various carborane-containing bioactive compounds as candidate nuclear receptor ligands.5–8) One derivative, 1-(4-hydroxyphenyl)-12-hydroxymethyl-p-carborane (BE120, 1), shows more potent estrogenic activity than the endogenous female hormone estrogen, estradiol (E2, 2, Fig. 1).1,9,10) Subsequently, many carboranyl phenol derivatives have been synthesized and their estrogen-related biological activities examined.11–18) Compounds that either induce or inhibit cellular estrogenic responses are used for estrogen replacement therapy after menopause or the treatment of breast cancer, respectively.19–21) Tissue-selective ligands, which act as agonists for bone metabolism and as antagonists for the female reproductive systems (known as selective estrogen receptor modulators, SERMs) are widely used for the treatment of osteoporosis.22)
The estrogen receptor subtypes ERα23) and ERβ24) are expressed in breast and uterus and in bone, colon, endothelial cells, bladder, and areas of the brain important for cognition, respectively. Most ER modulators are non-selective for ER subtypes, but it has been proposed that compounds with ER subtype selectivity would be useful as third-generation SERMs.25,26) Harris et al. showed that ERβ-selective ligands may be therapeutically useful to treat chronic intestinal and joint inflammation by modulating the immune response.27) Thus, ERβ-selective ligands are of interest as potential clinical candidates and also as probes for ERβ-related molecular biology.
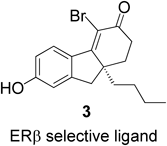
The X-ray crystal structures of receptor-ligand complexes of ERα and ERβ show that their binding pockets are quite similar.28) Binding of 2 to the ligand binding domains (LBDs) of the two ERs is illustrated in Fig. 2. The same amino acid residues are involved in hydrogen bonding with the two hydroxyl groups of E2 in the LBDs of ERα and ERβ, and thus E2 is accommodated equally well by the two LBDs. However, in the hydrophobic pocket of the LBDs, Leu384 and Met421 of ERα are substituted by Met336 and Ile373 in ERβ, respectively (Fig. 2). Compound 3 is an ERβ-selective ligand which was designed based the idea of decreasing the affinity for ERα by introducing a n-butyl group that interferes sterically with Met421 of ERα29) (Fig. 3).

We have synthesized a range of carboranyl phenol derivatives and have discovered various types of ER modulators, partial agonists, super-agonists, antagonists, and SERMs.11–18) In addition, our quantitative structure–activity relationship (QSAR) studies of very simple carboranyl phenols showed that the hydrophobicity of the carborane cage is significantly correlated with ERα-binding affinity.30,31) Although carboranyl phenol is a privileged structure as an ER ligand, little is known about the effect of aliphatic substituents on the carborane cage. Therefore, we designed and synthesized carboranyl phenol derivatives 4 and 5 with aliphatic substituents on the carborane cage for examination of their ERβ selectivity and estrogenic activities. From the viewpoints of ease of synthesis and the directions of the aliphatic substituents, we focused on o-carboranyl phenol and m-carboranyl bisphenol as being most relevant to drug design.
Synthesis of o-carboranyl phenol derivatives 4a–c is summarized in Chart 1. 4-Methoxyphenyl-o-carborane 6, which was synthesized by the literature procedure, was reacted with ethyl iodide or allyl bromide in the presence of NaH as a base to afford C-ethyl or C-allyl intermediates in 77% or 50% yield, respectively. Demethylation of the methoxy group of 7 with BBr3 afforded the target compounds 4a and b in 96% and 60% yields, respectively. The allyl group of compound 4b was reduced by catalytic hydrogenation using Pd/C to afford the corresponding C-n-propyl derivative 4c in 80% yield.
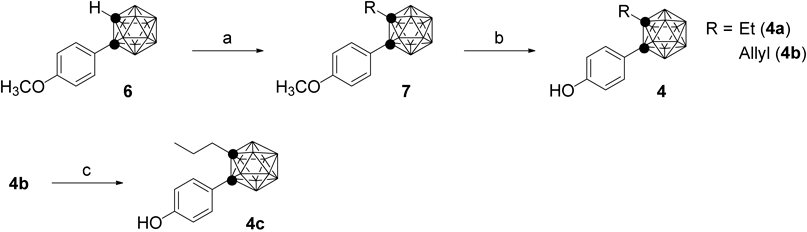
Reagents and conditions: a) NaH, ethyl iodide or allyl bromide, DMF, rt; b) BBr3, CH2Cl2, rt; c) Pd/C, H2, MeOH/AcOEt, rt.
Chart 2 summarizes the synthesis of m-carboranyl bisphenol derivatives 5a and b. 7-Iodo-m-carborane 8 was obtained by iodination of m-carborane.32) Grignard reagents, which were freshly prepared from Mg turnings and the corresponding halide, were reacted with 8 by Pd-catalyzed coupling reaction to afford B-allyl and B-n-butyl m-carborane in 99% and 61% yields, respectively.33) They were treated with n-butyl lithium to obtain the lithium salts, which were transformed into copper salts with Cu(I)Cl, and converted to the diaryl-m-carboranes 10a and b in 37% and 39% yields, respectively.34,35) B-n-Propyl derivative 5a was synthesized by catalytic hydrogenation of the allyl group of 10a, followed by deprotection of the phenol group with BBr3 in 45% yield over the 2 steps. Compound 5b was obtained by deprotection of the phenol group of 10b in 82% yield.

Reagents and conditions: a) Grignard reagents, Pd(PPh3)2Cl2, Cu(I)I, THF, reflux; b) i) n-BuLi, DME, rt, ii) Cu(I)Cl, pyridine, 4-iodoanisole, 100°C; c) Pd/C, H2, MeOH/AcOEt, rt; d) BBr3, CH2Cl2, rt.
The ER binding affinities of the synthesized compounds were evaluated by means of competitive ER binding assays using [2,4,6,7-3H]17β-estradiol and human recombinant ERα and ERβ.36) Table 1 summarized the relative binding affinity (RBA) values, estimated based on the RBA of estradiol 2, taken as 100. All the test compounds 4 and 5 bound to the ERs in a dose-dependent manner. Introduction of substituents onto the carborane cage decreased the binding affinity for both ERs. As the substituent was lengthened, the o-carboranyl phenol derivatives 4 showed a decrease of binding affinity for both ERs, but the decrease was more marked for ERα. Compound 4a showed weak binding affinity for ERα but its binding affinity for ERβ was higher than that of E2. This may suggest that ethyl group has an unfavorable steric interaction with the hydrophobic pocket of ERα, but not that of ERβ. The RBA values of allyl derivative 4b were decreased for both ERs. The allyl group is more polar than a n-propyl group and would be disfavored in a hydrophobic environment. Compound 4c showed a high RBA value with 7.4-fold selectivity for ERβ. These results show that introduction of an alkyl group onto the carborane cage markedly influences the ERβ selectivity. On the other hand, m-carborane bisphenol derivatives 5a and b showed little ER-subtype selectivity. We previously found that non-substituted m-carborane bisphenol had a similar RBA value to E2, while introduction of an alkylamino group into the second phenol group greatly decreased the binding affinity and changed the functional activity from ER agonist to ER antagonist.37) It seems that the phenol group is accommodated in a secondary hydrophobic pocket and forms hydrogen bonds in both ER LBDs.
| Compound | R | RBAa) | Selectivity ERβ/ERα | ||
|---|---|---|---|---|---|
| ERα | ERβ | ||||
| 4a | 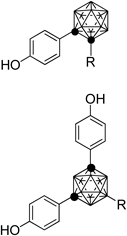 | Ethyl | 43.7 | 140.7 | 3.2 |
| 4b | Allyl | 9.3 | 44.9 | 4.8 | |
| 4c | n-Propyl | 11.8 | 87.0 | 7.4 | |
| 5a | n-Propyl | 27.6 | 21.7 | 0.8 | |
| 5b | n-Butyl | 32.4 | 43.5 | 1.3 | |
| E2 (1) | 100 | 100 | 1.0 | ||
a) Relative binding affinity with respect to that of estradiol 2 taken as 100%. All binding assays were performed in duplicate (n=2) and the numbers are average values.
A docking simulation of hERα (PDB: 1ERE) and hERβ (PDB: 2I0G) with the carborane containing ligands, 4c and 5b, was performed with an automatic docking program (GOLD 5.2).38) A docking mode of 4c in the hERβ LBD is shown in Fig. 4a. The highly hydrophobic o-carborane cage and n-propyl group are located in the hydrophobic pocket of the receptor, but the o-carborane cage of 4c is close to Ile373 of hERβ, contrary to our hypothesis (Fig. 4a). Interestingly, the arrangement of 4c in the LBDs of ERα is different from that in ERβ (Fig. 4b) and n-propyl group of 4c is close to Met421 in the hERα LBD (Fig. 4a). We suggested that the ERβ selectivity of 4c is caused by the steric repulsion between the n-propyl group and Met421 of hERα. A docking study of 5b with hERs showed that the second phenol group is accommodated in the secondary hydrophobic pocket of both ER LBDs (data not shown). The decrease of binding affinity and the poor selectivity of 5b for ERs may suggest that the direction of n-butyl group is not suitable for steric repulsion to occur with Met421 of ERα.

(a) Docking mode of 4c in the hERβ LBD (atom colors: H (white) B (green), C (gray), N (blue), O (red) and S (yellow)). The corresponding amino acid residues of hERα are colored orange. Both Ile373 of ERβ and Met421 of ERα are displayed with a ball and stick model. (b) The arrangements of 4c in the LBDs of ERα (brown) and ERβ (aqua).
Next, we evaluated the agonist and antagonist activities of the most ERβ-selective compound 4c by means of cell proliferation assay using the MCF-7 cell line, which shows estrogen-dependent growth.36) Compound 4c promoted cell proliferation in a dose-dependent manner and the EC50 value was 3.0×10−9 M (Fig. 5). When the maximum potency (Emax) of E2 was taken as 100%, that of 4c was 110%. Thus, compound 4c is simply a weaker ER full agonist.
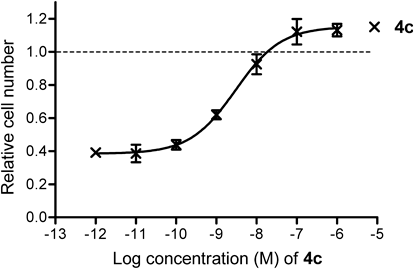
MCF-7 cells were incubated with 4c (1×10−6 to 1×10−12 m) for 4 d, and the results are shown as relative cell number, with the value for 0.1 nM estradiol taken as 1. Dashed line shows Emax of E2. Cell proliferation assays was performed in triplicate (n=3). Values are means±S.D. for separate experiments.
In conclusion, we examined the effect of introducing an aliphatic substituent into the carborane cage of o- or m-carboranyl phenol derivatives based on the structure of a well-known ERβ-selective ligand 3. Interestingly, the ERβ selectivity of o-carboranyl phenol derivatives 4 was enhanced as the length of the substituent was increased, whereas m-carboranyl phenol derivatives 5 showed little ERβ selectivity. The C-n-propyl o-carboranyl phenol 4c showed 7.4-fold ERβ selectivity in ER-binding assay and moderate estrogenic activity in MCF-7 cell proliferation assay. These results provide useful information on substituent effects to guide research aimed at discovery of selective carborane-containing estrogen modulators.
Melting points were determined with a Yanaco micro melting point apparatus and were not corrected. 1H- and 13C-NMR spectra were recorded with JEOL JNM-EX-270 and JNM-LA-400 spectrometers. Chemical shifts of 1H-NMR signals were referenced to tetramethylsilane (0.0 ppm) as an internal standard. Chemical shifts of 13C-NMR signals were referenced to residual 13C present in deuterated solvents. The splitting patterns are designated as follows: s (singlet), d (doublet), t (triplet), m (multiplet), q (quartet), and br (broad). Mass spectra were recorded on a JEOL JMS-DX-303 spectrometer. Column chromatography was carried out using Fuji Silysia BW80s and TLC was performed on Merck silica gel F254. Carboranes were purchased from Katchem s.r.o. Other chemicals were purchased from Aldrich or Tokyo Kasei Ltd. and used without further purification, or were prepared according to procedures described in the literature. All solvents were commercial products of reagent grade, and were used without further purification.
General Procedure of C-Alkylation of 6To a solution of 6 in dry N,N-dimethylformamide (DMF) was added NaH (1.2 eq) at 0°C under an argon (Ar) atmosphere, and the mixture was stirred for 30 min at the same temperature. Alkyl halide (1.2 eq) was added, and stirring was continued at room temperature for 2.5 h. The reaction mixture was poured onto ice and extracted with Et2O. The organic layer was washed with brine, dried over Na2SO4, and concentrated. The residue was purified by silica gel column chromatography with 1 : 20 AcOEt–n-hexane to give the corresponding C-alkylated compound.
1-(4-Methoxyphenyl)-2-ethyl-1,2-dicarba-closo-dodecaborane: Colorless solid; 77% yield; 1H-NMR (400 MHz, CDCl3) δ (ppm): 0.97 (t, J=7.7 Hz, 3H), 1.60–3.60 (br m, 10H), 1.85 (q, J=7.7 Hz, 2H), 3.82 (s, 3H), 6.87 (d, J=9.2 Hz, 2H), 7.53 (d, J=9.2, 2H); MS (electron ionization (EI)) m/z 278 (M+, 100%).
1-(4-Methoxyphenyl)-2-allyl-1,2-dicarba-closo-dodecaborane: Colorless solid; 50% yield; 1H-NMR (400 MHz, CDCl3) δ (ppm): 1.60–3.60 (br m, 10H), 2.53 (d, J=7.7 Hz, 2H), 3.83 (s, 3H), 4.73–4.79 (m, 1H), 5.03 (d, J=10.1 Hz, 1H), 5.56–5.68 (m, 1H), 6.88 (d, J=8.7 Hz, 2H), 7.54 (d, J=8.7, 2H); MS (EI) m/z 290 (M+, 100%).
General Procedure of Demethylation of 7To a solution of 6 in CH2Cl2 was added a solution of 1 M BBr3 (1.2 eq) in CH2Cl2 at −78°C, and the mixture was stirred at room temperature for 30 h, then poured onto ice and extracted with AcOEt. The organic layer was washed with brine, dried over Na2SO4, and concentrated. The residue was purified by silica gel column chromatography with 1 : 5 AcOEt–n-hexane to give the phenol derivative 4.
1-(4-Hydroxyphenyl)-2-ethyl-1,2-dicarba-closo-dodecaborane (4a): 96% yield; colorless needles (AcOEt–n-hexane); mp 106.5–108.0°C; 1H-NMR (400 MHz, CDCl3) δ (ppm): 0.97 (t, J=7.7 Hz, 3H), 1.60–3.60 (br m, 10H), 1.85 (q, J=7.7 Hz, 2H), 5.25 (br s, 1H), 6.81 (d, J=9.2 Hz, 2H), 7.50 (d, J=8.7, 2H). MS (EI) m/z 264 (M+, 100%); high resolution (HR)-MS Calcd for C10H20B10O: 264.2517, Found: 264.2521.
1-(4-Hydroxyphenyl)-2-allyl-1,2-dicarba-closo-dodecaborane (4b): 60% yield; colorless needles (CH2Cl2–n-hexane); mp 96.0–97.5°C; 1H-NMR (400 MHz, CDCl3) δ (ppm): 1.60–3.60 (br m, 10H), 2.54 (d, J=7.2 Hz, 2H), 4.72–4.79 (m, 1H), 5.03 (d, J=10.1 Hz, 1H), 5.34 (br s, 1H), 5.56–5.68 (m, 1H), 6.81 (d, J=8.7 Hz, 2H), 7.50 (d, J=8.7, 2H); MS (EI) m/z 276 (M+, 100%); HR-MS Calcd for C11H20B10O: 276.2517, Found: 276.2516.
1-(4-Hydroxyphenyl)-2-n-propyl-1,2-dicarba-closo-dodecaborane (4c): To a solution of 4b (25 mg) in AcOEt (5 mL) and MeOH (5 mL) was added Pd on carbon (5 mg) and the mixture was stirred at room temperature for 30 min under a hydrogen atmosphere. The mixture was filtered, the residue was washed with AcOEt, and the organic layer was concentrated. The residue was purified by silica gel column chromatography with 1 : 5 AcOEt–n-hexane to give 4c (21 mg, 80%) as colorless solid. Colorless oil; 1H-NMR (400 MHz, CDCl3) δ (ppm): 0.74 (t, J=7.2 Hz, 3H), 1.60–3.60 (br m, 10H), 1.41 (sext, J=7.7 Hz, 2H), 1.73 (t, J=8.7 Hz, 2H), 5.45 (br s, 1H), 6.82 (d, J=8.7 Hz, 2H), 7.49 (d, J=8.7, 2H); MS (EI) m/z 278 (M+, 100%); HR-MS Calcd for C11H22B10O: 278.2674, Found: 278.2678.
General Procedure of Synthesis of 9A solution of Grignard reagent (4 eq) was added dropwise to a stirred dry tetrahydrofuran (THF) solution (10 mL) of 8 (1.0 g, 3.70 mmol) at 0°C under an Ar atmosphere, and stirring was continued at room temperature for 30 min. Bis(triphenylphosphine)palladium(II) dichloride (260 mg, 0.37 mmol) and cupper(I) iodide (70.5 mg, 0.37 mmol) were added in one portion, and the reaction mixture was refluxed for 60 h. After removal of the solvent, Et2O was added to the residue and the excess Grignard reagent was destroyed by slow addition of 10% HCl aqueous solution. The organic phase was separated from the mixture, and the aqueous layer was extracted with Et2O. The combined organic phase was washed with water and brine, dried over Na2SO4, and concentrated. The residue was purified by column chromatography on silica gel with n-hexane to afford the corresponding 9.
9-Allyl-1,7-dicarba-closo-dodecaborane (9a): 99% yield; 1H-NMR (400 MHz, CDCl3) δ (ppm): 1.40–3.50 (br m, 9H), 1.78 (br s, 2H), 2.88 (br s, 2H), 4.75–4.90 (m, 2H), 5.86 (m, 1H); MS (EI) m/z 184 (M+, 100%).
9-n-Butyl-1,7-dicarba-closo-dodecaborane (9b): 61% yield; 1H-NMR (400 MHz, CDCl3) δ (ppm): 0.80–0.95 (m, 5H), 1.30–1.40 (m, 5H), 1.40–3.50 (br m, 9H), 2.85 (br s, 2H); MS (EI) m/z 200 (M+, 100%).
General Procedure of Synthesis of 10To a solution of 9 (350 mg) in 1,2-dimethoxyethane (DME) (3 mL) was added dropwise a solution of 2.66 M n-BuLi in hexane (2.2 eq) at 0°C under an Ar atmosphere, and the mixture was stirred for 30 min. Cu(I)Cl (2.2 eq) was added in one portion, and stirring was continued at room temperature for 1 h. Pyridine (15 eq) and p-iodoanisole (2 eq) were added in one portion, and the mixture was refluxed for 20 h, then cooled to room temperature, and diluted with AcOEt. Insoluble materials were filtered off through Celite. The filtrate was washed with 10% HCl aqueous solution, 10% Na2S2O3 aqueous solution, water and brine, dried over Na2SO4, and concentrated. The residue was purified by silica gel column chromatography with 1 : 30 to 1 : 20 AcOEt–n-hexane to give the corresponding 10.
1,7-Bis-(4-methoxyphenyl)-9-allyl-1,7-dicarba-closo-dodecaborane (10a): 37% yield; 1H-NMR (400 MHz, CDCl3) δ (ppm): 1.60–3.60 (br m, 9H), 1.86 (d, J=7.2 Hz, 2H), 3.76 (s, 6H), 4.80–4.93 (m, 2H), 5.91 (m, 1H), 6.75 (d, J=8.7 Hz, 4H), 7.36 (d, J=9.2 Hz, 4H); MS (EI) m/z 396 (M+, 100%).
1,7-Bis-(4-methoxyphenyl)-9-n-butyl-1,7-dicarba-closo-dodecaborane (10b): 39% yield; 1H-NMR (400 MHz, CDCl3) δ (ppm): 0.87–0.96 (m, 5H), 1.31–1.41 (m, 4H), 1.60–3.60 (br m, 9H), 3.78 (s, 6H), 6.76 (d, J=8.7 Hz, 4H), 7.37 (d, J=8.7 Hz, 4H); MS (EI) m/z 412 (M+, 100%).
1,7-Bis-(4-methoxyphenyl)-9-n-propyl-1,7-dicarba-closo-dodecaboraneThe title compound was prepared by the same method as described for the synthesis of 4c. 55% yield; 1H-NMR (400 MHz, CDCl3) δ (ppm): 0.92 (t, J=8.3 Hz, 2H), 0.97 (t, J=7.3 Hz, 3H), 1.43 (sext, J=7.8 Hz, 2H), 1.60–3.60 (br m, 9H), 3.77 (s, 6H), 6.76 (d, J=8.8 Hz, 4H), 7.37 (d, J=8.8 Hz, 4H).
1,7-Bis-(4-hydroxyphenyl)-9-n-propyl-1,7-dicarba-closo-dodecaborane (5a): 82% yield; colorless oil; 1H-NMR (400 MHz, CDCl3) δ (ppm): 0.91 (t, J=7.7 Hz, 2H), 0.96 (t, J=7.2 Hz, 3H), 1.42 (sext, J=7.2 Hz, 2H), 1.60–3.60 (br m, 9H), 5.32 (br s, 2H), 6.69 (d, J=8.7 Hz, 4H), 7.31 (d, J=8.7 Hz, 4H); MS (EI) m/z 370 (M+, 100%); HR-MS Calcd for C17H26B10O2: 370.2936, Found: 370.2943.
1,7-Bis-(4-hydroxyphenyl)-9-n-butyl-1,7-dicarba-closo-dodecaborane (5b): Quant; colorless oil; 1H-NMR (400 MHz, CDCl3) δ (ppm): 0.86–0.96 (m, 5H), 1.30–1.43 (m, 4H), 1.60–3.60 (br m, 9H), 5.82 (br s, 2H), 6.69 (d, J=8.7 Hz, 4H), 7.31 (d, J=8.7 Hz, 4H); MS (EI) m/z 384 (M+, 100%); HR-MS Calcd for C18H28B10O2: 384.3092, Found: 384.3094.
Competitive Binding Assay for ERsLigand binding to estrogen receptors α (ERα) and β (ERβ) was determined by the nitrocellulose filter binding assay method. ER (0.5 µg/tube, PanVera Co., Ltd.) was diluted with binding assay buffer (20 mM Tris–HCl pH 8.0, 0.3 M NaCl, 1 mM ethylenediaminetetraacetic acid (EDTA) pH 8.0, 10 mM 2-mercaptoethanol, 0.2 mM phenylmethylsulfonyl fluoride) and incubated with 4 nM [6,7-3H]17β-estradiol in the presence or absence of an unlabeled competitor at 4°C for 18 h. The incubation mixture was absorbed by suction onto a nitrocellulose membrane that had been soaked in binding assay buffer. The membrane was washed twice with buffer (20 mM Tris–HCl pH 8.0, 0.15 M NaCl) and then with 25% ethanol in distilled water. Radioactivity that remained on the membrane was measured in Atomlight (NEN) by using a liquid scintillation counter.
Cell CultureAt 80% confluence, cells of the human breast adenocarcinoma cell line MCF-7 were trypsinized from the maintenance dish with 0.25% trypsin–EDTA and collected by centrifugation (4°C, 1500 rpm, 5 min). The supernatant was removed, 1 mL of Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 IU/mL penicillin and 100 mg/mL streptomycin was added, and cell aggregates were broken up by thorough pipetting. Cells were seeded in a dish at a concentration of 4×104 cell/mL or 8×104 cell/mL, and cultivated at 37°C in a 5% CO2 humidified incubator. Cells were routinely cultivated 2 d or 3 d later.
Cell Proliferation AssayCells of the human breast adenocarcinoma line MCF-7 were routinely cultivated in DMEM supplemented with 10% FBS, 100 IU/mL penicillin and 100 mg/mL streptomycin at 37°C in an incubator under an atmosphere of 5% CO2 in air. On the day before assay, MCF-7 cells were switched to DMEM (low glucose phenol red-free supplemented with 5% sFBS, 100 IU/mL penicillin and 100 mg/mL streptomycin). Cells were trypsinized from the maintenance dish into phenol red-free trypsin–EDTA and seeded in a 96-well plate at a density of 2×103 cells per final volume of 100 µL DMEM supplemented with 5% sFBS, 100 IU/mL penicillin and 100 mg/mL streptomycin. After 24 h, the medium was replaced with 90 µL of fresh DMEM and 10 µL of drug solution, supplemented with serial dilutions of 4-OH-Tam or DMSO as the dilution control in the presence or absence of 1×10−11 M E2, was added to triplicate microcultures. Cells were incubated for 4 d, and medium with 4-OH-Tam in the presence or absence of 1×10−11 M E2 was replaced once after 3 d. At the end of the incubation time, proliferation was evaluated by using a cell counting kit. WST-8 (10 µM) was added to microcultures and cells were incubated for 2 h. The absorbance at 450 nm was measured. This parameter is related to the number of living cells in the culture.
Docking StudyThree-dimensional (3D) structures of protein-ligand complexes were predicted by GOLD 5.2 software with default settings.38) The 3D structure of human ERα and β used in this study was retrieved from the Protein DataBank (PDB) (PDB ID: 1ERE and 2I0G, respectively). Missing hydrogen atoms in the crystal structure were computationally added by Hermes.39) The center of the active site was defined as the center of the ligand in 1ERE and 2I0G for ERα and β, respectively, and the active site radius was set to 10.0 Å. The structural optimizations of ligands were carried out by B3LYP/6-31G(d,p) using Gaussian 09, Revision C.01.40) The cavities of ER were detected by Q-SiteFinder.41) PDB entries 1ERE and 2I0G were used as 3D structures of ERα and β similar to the docking study. Ten sites were output for each protein and the sites were ranked by volume (default setting).
This research was supported by a Grant-in-Aid from the Strategic Research Program for Private Universities (2010–2014), a Grant-in-Aid for Scientific Research (B) (No. 20390035) and a Grant-in-Aid for Young Scientists (B) (No. 21790116) from the Ministry of Education, Culture, Sports, Science and Technology of Japan.