Notes
New Nitrogenous Bisabolene-Type Sesquiterpenes from a Formosan Sponge Axinyssa sp.
2014 年 62 巻 4 号 p. 392-394

詳細
2014 年 62 巻 4 号 p. 392-394
Chemical investigation of a Formosan sponge Axinyssa sp. has led to the isolation of five nitrogenous bisabolene-type sesquiterpenes 1–5, including two new compounds axinysalines A (1) and B (2). The structures of new compounds were elucidated by analysis of high resolution (HR)-MS and two dimensional (2D)-NMR spectra and comparison of its NMR data with those of known analogues. Compound 1 exhibited moderate to weak cytotoxicity against Molt 4 and K562 cancer cell lines.
Studies in the chemistry of marine natural products, the marine sponges have emerged as one of the most prolific sources for discovery of novel secondary metabolites.1) Previous chemical investigations on sponges of the genus Axinyssa (order Halichondrida, family Halichondriidae) have led to the isolation and identification of various sesquiterpenes containing unusual nitrogenous functional groups as an isocyano, isothiocyanate, thiocyanate, bisabolene and formamide group.2–15) Some of these N-containing compounds have been found to possess several kinds of biological activities, such as antithelmintic,2) antimicrobial,4) antimalarial,6) antifouling,11) cytotoxic activities12) and lethality for brine shrimp.10) Our investigation of the chemical constituents of a sponge Axinyssa sp. yielded two new nitrogenous bisabolene-type sesquiterpenes, axinysalines A (1) and B (2), along with three known sesquiterpenes. The structures of 1 and 2 were established by detailed spectroscopic analysis and comparison of their NMR data with those of known analogues. The known sesquiterpenes were readily identified as 3-isocyanotheonellin (3),16) 3-isocyano-7,8-epoxy-α-bisabolane (4)7) and 3-formamidotheonellin (5),17) by comparison of their spectral data with those reported in the literature. The cytotoxicity of metabolites 1–5 against the six cancer cell lines, human erythromyeloblastoid leukemia (K562), human T lymphoblast, acute lymphoblastic leukemia (Molt 4), human colon adenocarcinoma (HCT-116 and DLD-1), and human breast carcinoma (MDA-MB-231 and T47D) was studied.
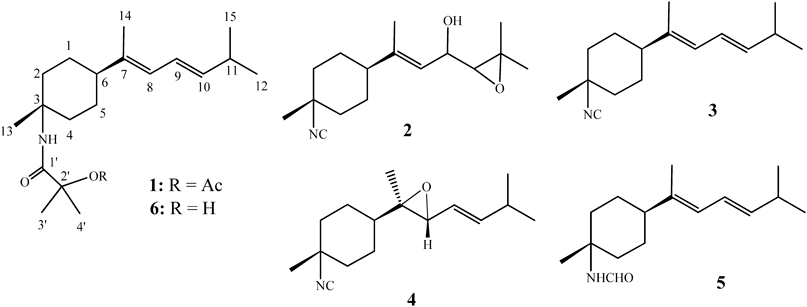
Compound 1, named axinysaline A, was isolated as an optically active, colorless oil. The molecular formula was determined to be C21H35NO3 by high resolution-electrospray ionization (HR-ESI)-MS (m/z 372.2515, C21H35NO3Na) and NMR data, which corresponded to five degrees of unsaturation. The IR spectrum of 1 showed absorption bands at 3375, 1679 and 1746 cm−1, suggesting the presence of an amide CO group and an ester CO group, which were supported by 13C-NMR chemical shifts at δ 171.9 and 169.2 ppm, respectively. The 13C-NMR and distortionless enhancement by polarization transfer (DEPT) spectral data of 1 showed the presence of 21 carbons (Table 1): seven methyls, four sp3 methylenes, two sp3 methines, three sp2 methines, and two sp3 quaternary carbons. The remaining three signals appearing in the lower field region of the spectrum are due to the quaternary carbons of one olefinic carbon (δ 140.2), one amide carbonyl and one ester carbonyl. From the 1H-NMR spectrum of 1, the resonances of three olefinic protons (δH 6.22, dd, J=14.5, 11.0 Hz; 5.81, d, J=11.0 Hz; 5.59 dd, J=15.5, 7.0 Hz) were observed. Moreover, analysis of the 1H-NMR spectrum suggested that one of the seven methyl groups were due to acetate moiety which occurred at δH 2.07 (s). Four of the six remaining methyl groups appeared as singlet at δH 1.73, 3H; 1.59, 6H; 1.39, 3H, while two methyls showed doublet at δH 1.02, 6H, J=6.5 Hz). The planar structure and all of the 1H and 13C chemical shifts of 1 were elucidated by two dimensional (2D)-NMR spectroscopic analysis, in particular 1H–1H correlation spectroscopy (COSY) and heteronuclear multiple bond connectivity (HMBC) experiments (Fig. 1). The relative stereochemistry of 1 was mostly confirmed to be the same as those of 3 and 5 by comparison of the H and C chemical shifts with published values. Moreover, the relative stereochemistry was also confirmed by observed nuclear Overhauser effect spectroscopy (NOESY) correlations (Fig. 2). Furthermore, by comparison of the NMR data of 1 with the known compound (6),13) it was found that the 1H- and 13C-NMR data of 1 were very similar to those of 6, with the difference that 1 contains one more acetyl group relative to 6. The chemical shift of H3-3′ and H3-4′ in 1 (δ 1.59) was shifted upfield (δ 1.42) in 6, suggesting that 1 is the 2′-acetyl derivative of 6. On the basis of above analysis, the structure of 1 was established.
| Position | 1 | 2 | ||
|---|---|---|---|---|
| δH (J in Hz)a) | δC (mult.)b) | δH (J in Hz)a) | δC (mult.)b) | |
| 1 | 1.63 m; 1.44 m | 27.3 (CH2) c) | 1.41 m; 1.75 m | 26.5 (CH2) |
| 2 | 2.02 m; 1.65 m | 36.6 (CH2) | 1.84 m; 1.96 m | 38.2 (CH2) |
| 3 | 53.2 (C) | 56.6 (C) | ||
| 4 | 2.02 m; 1.65 m | 36.6 (CH2) | 1.84 m; 1.96 m | 38.2 (CH2) |
| 5 | 1.63 m; 1.44 m | 27.3 (CH2) | 1.41 m; 1.75 m | 26.4 (CH2) |
| 6 | 1.93 m | 46.4 (CH) | 2.00 m | 44.7 (CH) |
| 7 | 140.2 (C) | 143.7 (C) | ||
| 8 | 5.81 d (11.0) | 123.3 (CH) | 5.30 br d (8.0) | 121.9 (CH) |
| 9 | 6.22 dd (15.0; 11.0) | 123.5 (CH) | 4.23 dd (8.0;7.5) | 67.6 (CH) |
| 10 | 5.59 dd (15.0; 7.0) | 140.2 (CH) | 2.80 d (7.5) | 67.3 (CH) |
| 11 | 2.34 m | 31.4 (CH) | 59.9 (C) | |
| 12 | 1.02 d (6.5) | 22.5 (CH3) | 1.33 d (8.0) | 24.9 (CH3) |
| 13 | 1.39 s | 21.9 (CH3) | 1.45 s | 25.0 (CH3) |
| 14 | 1.73 s | 15.0 (CH3) | 1.69 s | 15.7 (CH3) |
| 15 | 1.02 d (6.5) | 22.5 (CH3) | 1.31 d (8.0) | 19.5 (CH3) |
| 1′ | 171.9 (C) | |||
| 2′ | 81.7 (C) | |||
| 3′ | 1.59 s | 24.3 (CH3) | ||
| 4′ | 1.59 s | 24.3 (CH3) | ||
| OAc | 169.2 (C) | |||
| 2.07 s | 22.0 (CH3) | |||
| NH | 5.79 s | |||
| NC | 152.3 (C) | |||
a) 500 MHz in CDCl3. b) 125 MHz in CDCl3. c) Numbers of attached protons were deduced by DEPT experiments.


Axinysaline B (2) was obtained as a colorless oil and showed a [M+Na]+ ion peak in the HR-ESI-MS (m/z 286.1783 [M+Na]+) and NMR spectroscopic data (Table 1) corresponding to the molecular formula C16H25NO2. By analysis of 2D-NMR spectra (Fig. 1), including 1H–1H COSY, heteronuclear multiple quantum coherence (HMQC) and HMBC, compound 2 was shown to possess the same molecular framework as those of 3 and 4. Moreover, the correlations observed in the NOESY spectrum of 2 also showed that 2 possessed the same relative configurations at C-3 and C-6, as those of 1 and 3–5.
The cytotoxicity of metabolites 1–5 against the growth of human erythromyeloblastoid leukemia (K562), human T lymphoblast, acute lymphoblastic leukemia (Molt 4), human colon adenocarcinoma (HCT-116 and DLD-1), and human breast carcinoma (MDA-MB-231 and T47D) was studied. The results showed that compounds 2–5 are not cytotoxicity (IC50’s >20 µg/mL) toward the above cancer cells. Compound 1 exhibited moderate to weak cytotoxicity against Molt 4 and K562 cancer cell lines with IC50 values of 14.3 and 4.7 µg/mL, respectively.
Optical rotation values were measured with a Jasco P-1010 digital polarimeter. IR spectra were recorded on a Varian Digilab FTS 1000 Fourier transform infrared spectrophotometer. The NMR spectra were recorded on a Varian Unity INOVA 500 FT-NMR instrument at 500 MHz for 1H-NMR and 125 MHz for 13C-NMR, respectively, in CDCl3. ESI-MS were obtained with a Bruker APEX II mass spectrometer. Gravity column chromatography was performed on silica gel (230–400 mesh, Merck). TLC was carried out on precoated Kieselgel 60 F254 (0.2 mm, Merck) and spots were visualized by spraying with 10% H2SO4 solution followed by heating. High-performance liquid chromatography (HPLC) was performed using a system comprised of a Hitachi L-7100 pump and a Rheodyne 7725 injection port. A preparative normal phase column (Hibar 250×21.2 mm, Supelco, silica gel 60, 5 µm) was used for HPLC.
The specimen of Axinyssa sp. was collected by scuba divers at a depth of 5–10 m from coral reefs off the coast of Pingtung, Taiwan, in April, 2012. A voucher specimen was deposited in the National Museum of Marine Biology and Aquarium, Taiwan (specimen no. 2012SP-1).
The sponge (590 g, fresh wt) stored frozen and then freeze dried. The freeze-dried material (110 g) was minced and extracted exhaustively with EtOAc. The EtOAc extract was evaporated to yield a residue (1.8 g), which was subjected to open column chromatography on silica gel eluting with n-hexane–EtOAc gradient and EtOAc–acetone gradient, to give 19 fractions. Fraction 3 eluted with n-hexane–EtOAc (20 : 1) and was chromatographed by normal phase HPLC using n-hexane–acetone (100 : 1) to afford 3 (387 mg, 21.5% dry wt of extract). Fraction 4 eluted with n-hexane–EtOAc (10 : 1) and was subjected to normal phase HPLC using n-hexane–acetone (50 : 1) to afford 7 subfractions (A1–A7). Subfraction A6 was separated by normal phase HPLC using n-hexane–acetone (50 : 1) to afford 4 (3.9 mg, 0.22% dry wt of extract). Fraction 7 eluted with n-hexane–EtOAc (5 : 1) to n-hexane–EtOAc (3 : 1) and was subjected to normal phase HPLC using n-hexane–acetone (5 : 1) to afford 13 subfractions (B1–B13). Subfraction B13 was separated by normal phase HPLC using n-hexane–acetone (10 : 1) to afford 2 (3.6 mg, 0.20% dry wt of extract). Fraction 10 eluted with n-hexane–EtOAc (2 : 1) and was subjected to normal phase HPLC using n-hexane–acetone (5 : 1) to afford 22 subfractions (C1–C22). Subfraction C21 was separated by normal phase HPLC using n-hexane–acetone (2 : 1) to afford 5 (8.2 mg, 0.47% dry wt of extract). Fraction 12 eluted with n-hexane–EtOAc (1 : 1) and was chromatographed by normal phase HPLC using n-hexane–acetone (3 : 1) to afford 28 subfractions (D1–D28). Subfraction D1 was separated by normal phase HPLC using n-hexane–acetone (2 : 1) to afford 1 (2.2 mg, 0.12% dry wt of extract).
Axinysaline A (1): Colorless oil; [α]D25 +1.09 (c=0.18, CHCl3); IR (neat) νmax 3375, 2932, 1746, 1679, 1148 cm−1; 13C- and 1H-NMR data, see Table 1; ESI-MS m/z 372 [M+Na]+; HR-ESI-MS m/z 372.2515 [M+Na]+ (Calcd for [C21H35NO3Na], 372.2513).
Axinysaline B (2): Colorless oil; [α]D25 −1.54 (c=0.11, CHCl3); IR (neat) νmax 3401, 2933, 2133, 1671, 1382 cm−1; 13C- and 1H-NMR data, see Table 1; ESI-MS m/z 286 [M+Na]+; HR-ESI-MS m/z 286.1783 [M+Na]+ (Calcd for [C16H25NO2Na], 286.1785).
Cell lines were purchased from the American Type Culture Collection (ATCC). Cytotoxicity assays of the compounds 1–5 were performed using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) colorimetric method. As a positive control, we employed doxorubicin, which exhibited cytotoxicity to K562, Molt 4, HCT-116, DLD-1, T47D and MDA-MB-231 cells with IC50 values of 0.17, 0.01, 0.56, 0.43, 0.99 and 0.09 µg/mL, respectively.18,19)
This work was supported by Grants from the National Museum of Marine Biology and Aquarium; the Division of Marine Biotechnology, Asia-Pacific Ocean Research Center, National Sun Yat-sen University and the National Science Council (Grant No. NSC 102-2325-B-291-001), Taiwan, awarded to J.-H. Su.