Experimental
General MethodsMelting points were determined on a Yanagimoto micro melting point apparatus or Büchi B-545 and are uncorrected. Proton and carbon nuclear magnetic resonance (NMR) spectra were recorded on a Varian Mercury-300 or a Bruker AV-300M spectrometer. Chemical shifts are given in δ values (ppm) using tetramethylsilane as the internal standard. Coupling constants (J) are reported in hertz (Hz). Spectral splitting patterns are designated as follows: s, singlet; br, broad; d, doublet; t, triplet; m, multiplet. Fourier transform infrared (FT-IR) spectra were made on Thermo 5700 Nicolet sprectrometer and were expressed in wave number (cm−1) using attenuated total reflectance (ATR) accessory. Microwave was irradiated with CEM Discover microwave focused chemical synthesis reactor. TLC analyses were carried out on Merck Kieselgel 60 F254 plates or FUJI SILYSIA Chemical Ltd. Chromatorex NH-TLC plates. Silica gel column chromatography was performed using Merck 0.063–0.200 mm silica gel 60, and FUJI SILYSIA Chemical Ltd. 100–200 mesh Chromatorex NH silica DM1020. Elemental analyses and high resolution (HR)-MS analyses were carried out by Takeda Analytical Research Laboratories, Ltd.
7-Chloro-2,3-dimethyl-1H-pyrrolo[2,3-c]pyridine (4)A solution of 2-chloro-3-nitropyridine (3) (3.5 g, 22 mmol) in tetrahydrofuran (THF) (100 mL) was cooled to −78°C, and a 0.5 M 1-methyl-1-propenylmagnesium bromide in THF (100 mL, 50 mmol) was added. The reaction mixture was stirred at −20°C for 18 h, warmed to room temperature, and concentrated under reduced pressure to a liquid amount of about 40 mL. Twenty percent aqueous ammonium chloride solution (200 mL) was added, and the mixture was extracted with EtOAc. The extract was washed with saturated brine, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (eluent: hexane–EtOAc=1 : 1). The collected fraction was concentrated and unreacted compound 3 was filtered off. The mother liquid was concentrated and the residue was washed with isopropyl ether (IPE) to give the title compound as a pale-yellow solid (580 mg, 3.2 mmol, 15%). mp 170–171°C. 1H-NMR (CDCl3) δ: 2.21 (3H, s), 2.44 (3H, s), 7.30 (1H, d, J=5.7 Hz), 7.98 (1H, d, J=5.7 Hz), 8.20 (1H, br).
N-Benzyl-2,3-dimethyl-1H-pyrrolo[2,3-c]pyridine-7-amine (1)A mixture of 4 (200 mg, 1.1 mmol) and benzylamine (1 mL, 9.2 mmol) was stirred at 180°C for 30 min under microwave irradiation. The mixture was cooled to room temperature (rt). A saturated aqueous sodium hydrogencarbonate solution was added to the reaction mixture which was then extracted with EtOAc. The extract was washed successively with a saturated aqueous sodium hydrogencarbonate solution, and brine, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (eluent: hexane–EtOAc=2 : 1) and the obtained solid was crystallized from IPE to give the title compound as a colorless solid (154 mg, 0.61 mmol, 56%). mp 180–182°C. 1H-NMR (CDCl3) δ: 2.17 (3H, s), 2.29 (3H, s), 4.51 (1H, br), 4.67 (2H, d, J=5.2 Hz), 6.85 (1H, d, J=5.2 Hz), 7.20–7.40 (5H, m), 7.78 (1H, d, J=5.2 Hz), 8.30 (1H, br). 13C-NMR (CDCl3) δ: 8.5, 11.4, 46.1, 105.2, 107.8, 119.4, 126.9, 127.6, 128.3, 133.3, 133.8, 135.6, 139.4, 145.2. IR (ATR) cm−1: 1632, 1570, 1555, 1476, 1452, 1406. Anal. Calcd for C16H17N3: C, 76.46; H, 6.82; N, 16.72. Found: C, 76.51; H, 6.94; N, 16.55.
Following compounds 5a and 5c–g were prepared in the similar manner with compound 1.
N-Benzyl-N,2,3-trimethyl-1H-pyrrolo[2,3-c]pyridine-7-amine (5a): Colorless solid (79%). mp 106–107°C. 1H-NMR (CDCl3) δ: 2.16 (3H, s), 2.24 (3H, s), 3.12 (3H, s), 4.69 (2H, s), 6.93–6.95 (1H, m), 7.26–7.46 (5H, m), 7.64 (1H, br s), 7.88 (1H, d, J=5.4 Hz). 13C-NMR (CDCl3) δ: 8.4, 11.6, 38.0, 56.3, 106.6, 107.7, 121.5, 127.1, 127.3, 128.9, 132.7, 135.4, 136.5, 139.3, 148.2. IR (ATR) cm−1: 1552, 1494, 1451, 1392. Anal. Calcd for C17H19N3: C, 76.95; H, 7.22; N, 15.84. Found: C, 76.82; H, 7.13; N, 15.82.
2,3-Dimethyl-N-(2-methylbenzyl)-1H-pyrrolo[2,3-c]pyridine-7-amine (5c): Colorless solid (90%). mp 225–226°C. 1H-NMR (CDCl3) δ: 2.16 (3H, s), 2.24 (3H, s), 2.28 (3H, s), 4.37 (1H, br), 4.60 (2H, d, J=4.8 Hz), 6.82 (1H, d, J=5.7 Hz), 7.01–7.14 (3H, m), 7.20–7.25 (1H, m), 7.75 (1H, d, J=5.7 Hz), 8.68 (1H, br). 13C-NMR (CDCl3) δ: 8.5, 11.5, 18.8, 44.2, 105.2, 108.0, 119.4, 125.9, 127.3, 128.6, 130.2, 132.9, 133.8, 136.0, 136.7, 137.3, 145.0. IR (ATR) cm−1: 1633, 1570, 1556, 1468, 1407. Anal. Calcd for C17H19N3: C, 76.95; H, 7.22; N, 15.84. Found: C, 76.84; H, 7.16; N, 15.86.
N-(2,6-Dimethylbenzyl)-2,3-dimethyl-1H-pyrrolo[2,3-c]pyridine-7-amine (5d): Colorless solid (84%). mp 279–280°C. 1H-NMR (CDCl3) δ: 2.17 (3H, s), 2.32 (3H, s), 2.34 (6H, s), 3.91 (1H, br), 4.66 (2H, d, J=4.2 Hz), 6.83 (1H, d, J=5.7 Hz), 6.98–7.01 (2H, m), 7.05–7.10 (1H, m), 7.80 (1H, d, J=5.7 Hz), 8.19 (1H, br). 13C-NMR (CDCl3) δ: 8.5, 11.6, 19.6, 40.5, 105.1, 108.2, 119.2, 127.5, 128.2, 132.5, 133.6, 135.5, 136.3, 137.8, 145.0. IR (ATR) cm−1: 1631, 1571, 1556, 1468, 1407. Anal. Calcd for C18H21N3: C, 77.38; H, 7.58; N, 15.04. Found: C, 77.22; H, 7.44; N, 15.02.
N-(4-Fluoro-2-methylbenzyl)-2,3-dimethyl-1H-pyrrolo[2,3-c]pyridine-7-amine (5e): Colorless solid (48%). mp 215–218°C. 1H-NMR (CDCl3) δ: 2.17 (3H, s), 2.18 (3H, s), 2.29 (3H, s), 4.45 (1H, br s), 4.52–5.54 (2H, m), 6.62–6.75 (2H, m), 6.83 (1H, d, J=5.7 Hz), 7.08–7.12 (1H, m), 7.72 (1H, d, J=5.7 Hz), 9.01 (1H, br s). 13C-NMR (CDCl3) δ: 8.5, 11.5, 18.9, 43.5, 105.3, 108.1, 112.1, 112.4, 116.6, 116.9, 119.2, 129.8, 130.0, 132.68, 132.73, 133.3, 133.7, 135.4, 139.0, 139.1, 144.8, 160.3, 163.5. IR (ATR) cm−1: 1635, 1569, 1554, 1470, 1412. Anal. Calcd for C17H18N3F: C, 72.06; H, 6.40; N, 14.83. Found: C, 71.85; H, 6.40; N, 14.46.
N-(2,3-Dihydro-1H-inden-1-yl)-2,3-dimethyl-1H-pyrrolo[2,3-c]pyridine-7-amine (5f): Colorless solid (46%). mp 245–247°C. 1H-NMR (CDCl3) δ: 1.62–1.92 (1H, m), 2.17 (3H, s), 2.32 (3H, s), 2.58–2.69 (1H, m), 2.78–2.98 (2H, m), 4.44 (1H, br), 5.72–5.79 (1H, m), 6.89 (1H, d, J=5.9 Hz), 7.06–7.11 (1H, m), 7.15–7.28 (3H, m), 7.78 (1H, d, J=5.9 Hz), 8.30 (1H, br). 13C-NMR (CDCl3) δ: 8.5, 11.6, 30.2, 34.6, 56.5, 105.2, 108.2, 119.4, 124.3, 124.6, 124.7, 126.4, 127.5, 132.9, 133.9, 136.1, 143.7, 144.7. IR (ATR) cm−1: 1633, 1572, 1555, 1471, 1406. Anal. Calcd for C18H19N3: C, 77.95; H, 6.90; N, 15.15. Found: C, 77.80; H, 6.86; N, 15.10.
2,3-Dimethyl-N-(1,2,3,4-tetrahydronaphthalen-1-yl)-1H-pyrrolo[2,3-c]pyridine-7-amine (5g): Colorless solid (34%). mp 248–250°C. 1H-NMR (CDCl3) δ: 1.76–1.86 (2H, m), 1.95–2.06 (2H, m), 2.17 (3H, s), 2.32 (3H, s), 2.72–2.76 (2H, m), 4.35 (1H, br), 5.43–5.49 (1H, m), 6.81 (1H, d, J=5.9 Hz), 7.03–7.16 (3H, m), 7.34–7.37 (1H, m), 7.77 (1H, d, J=5.9 Hz), 8.18 (1H, br). 13C-NMR (CDCl3) δ: 8.5, 11.6, 19.6, 29.3, 29.9, 48.6, 104.9, 108.2, 119.2, 126.0, 126.8, 128.9, 129.4, 132.7, 133.9, 136.2, 137.7, 138.4, 144.4. IR (ATR) cm−1: 1632, 1571, 1555, 1470, 1406. Anal. Calcd for C19H21N3: C, 78.32; H, 7.26; N, 14.42. Found: C, 78.08; H, 7.31; N, 14.47.
N-[7-(2,3-Dimethyl-1H-pyrrolo[2,3-c]pyridyl)]benzamide (5b)To a solution of 4 (306 mg, 1.7 mmol) in toluene (20 mL) were added Tris(dibenzylideneacetone) dipalladium (15.8 mg, 0.017 mmol), 9,9-dimethyl-4,5-bis(diphenylphosphino)xanthine (30.1 mg, 0.052 mmol), cesium carbonate (775 mg, 2.4 mmol) and benzamide (291 mg, 2.4 mmol), and the reaction mixture was stirred under an argon atmosphere at 120°C for 14 h. After cooling to room temperature, a saturated aqueous sodium hydrogencarbonate solution was added to the reaction mixture and the mixture was extracted with EtOAc. The extract was washed with a saturated aqueous sodium hydrogencarbonate solution, water and saturated brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (eluent: hexane–EtOAc=3 : 1) to give the title compound as colorless crystals (117 mg, 0.44 mmol, 26%). mp 163–165°C. 1H-NMR (CDCl3) δ: 2.22 (3H, s), 2.43 (3H, s), 7.20 (1H, d, J=5.7 Hz), 7.45–7.53 (2H, m), 7.54–7.62 (1H, m), 7.78 (1H, d, J=5.7 Hz), 7.97–8.05 (2H, m), 9.04 (1H, br s), 10.50 (1H, br s). 13C-NMR (CDCl3) δ: 8.4, 11.8, 107.4, 110.3, 122.0, 127.7, 128.7, 132.2, 133.8, 134.4, 136.0, 136.9, 137.3, 167.3. IR (ATR) cm−1: 1663, 1645, 1581, 1520, 1489, 1331. Electrospray ionization (ESI)-HR-MS m/z: 266.1287 (Calcd for C16H15N3O2S: 266.1288 [M+H]+).
7-Chloro-1,2,3-trimethyl-1H-pyrrolo[2,3-c]pyridine (6a)Sodium hydride (60% in oil, 329 mg, 8.2 mmol) was washed twice with hexane and suspended in N,N-dimethylformamide (DMF) (15 mL). A solution of 4 (1.19 g, 6.6 mmol) in DMF (5 mL) was added dropwise at 0°C. After stirring at the same temperature for 15 min, a solution of iodomethane (0.50 mL, 8.0 mmol) in DMF (5 mL) was added dropwise at 0°C and the mixture was stirred at room temperature for 14 h. Saturated aqueous sodium hydrogencarbonate solution was added to the reaction mixture and the mixture was extracted with EtOAc. The extract was washed with saturated aqueous sodium hydrogencarbonate solution, water and saturated brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. The residue was crystallized from IPE to give the title compound as colorless crystals (0.93 g, 4.8 mmol, 72%). mp 126–127°C. 1H-NMR (CDCl3) δ: 2.22 (3H, s), 2.37 (3H, s), 4.04 (3H, s), 7.28 (1H, d, J=5.4 Hz), 7.91 (1H, d, J=5.4 Hz).
Following compounds 6b–d were prepared in the similar manner with compound 6a.
7-Chloro-1-ethyl-2,3-dimethyl-1H-pyrrolo[2,3-c]pyridine (6b): Colorless crystals (73%). mp 65–67°C. 1H-NMR (CDCl3) δ: 1.36 (3H, t, J=7.2 Hz), 2.21 (3H, s), 2.37 (3H, s), 4.51 (2H, q, J=7.2 Hz), 7.28 (1H, d, J=5.4 Hz), 7.91 (1H, d, J=5.4 Hz).
7-Chloro-2,3-dimethyl-1-propyl-1H-pyrrolo[2,3-c]pyridine (6c): Colorless crystals (89%). mp 72–73°C. 1H-NMR (CDCl3) δ: 0.97 (3H, t, J=6.9 Hz), 1.71–1.83 (2H, m), 2.21 (3H, s), 2.37 (3H, s), 4.35–4.41 (2H, m), 7.27 (1H, d, J=5.1 Hz), 7.91 (1H, d, J=5.1 Hz).
7-Chloro-1-isobutyl-2,3-dimethyl-1H-pyrrolo[2,3-c]pyridine (6d): Colorless crystals (83%). mp 56–57°C. 1H-NMR (CDCl3) δ: 0.90 (6H, d, J=6.6 Hz), 2.21–2.30 (m, 1H), 2.45 (3H, d, J=0.9 Hz), 4.23 (2H, br d, J=6.9 Hz), 6.29 (1H, d, J=0.9 Hz), 7.31 (1H, d, J=5.4 Hz), 7.92 (1H, d, J=5.4 Hz).
Following compounds 2a and 7a–e were prepared in the similar manner with compound 1.
N-Benzyl-2,3-dimethyl-1-propyl-1H-pyrrolo[2,3-c]pyridine-7-amine (2a): Pale-yellow crystals (33%). mp 51–52°C. 1H-NMR (CDCl3) δ: 0.83 (3H, t, J=7.5 Hz), 1.67–1.80 (2H, m), 2.17 (3H, s), 2.29 (3H, s), 4.04–4.09 (2H, m), 4.50 (1H, br), 4.74 (2H, d, J=5.4 Hz), 6.85 (1H, d, J=5.7 Hz), 7.26–7.37 (3H, m), 7.41–7.45 (2H, m), 7.77 (1H, d, J=5.7 Hz). 13C-NMR (CDCl3) δ: 8.9, 10.3, 10.9, 25.9, 46.5, 47.0, 105.5, 107.3, 120.6, 127.2, 127.9, 128.6, 133.9, 134.9, 135.9, 140.2, 145.3. IR (ATR) cm−1: 1607, 1554, 1473, 1368. Anal. Calcd for C19H23N3: C, 77.78; H, 7.90; N, 14.32. Found: C, 77.69; H, 7.83; N, 14.43.
N-Benzyl-1,2,3-trimethyl-1H-pyrrolo[2,3-c]pyridine-7-amine (7a): Colorless solid (56%). mp 112–113°C. 1H-NMR (CDCl3) δ: 2.17 (3H, s), 2.29 (3H, s), 3.90 (3H, s), 4.67 (1H, br d, J=5.4 Hz), 4.74 (2H, d, J=5.4 Hz), 6.84 (1H, d, J=5.7 Hz), 7.26–7.37 (3H, m), 7.42–7.57 (2H, m), 7.75 (1H, d, J=5.7 Hz). 13C-NMR (CDCl3) δ: 8.9, 10.3, 32.5, 46.4, 105.6, 107.1, 121.4, 127.1, 127.9, 128.6, 133.5, 135.2, 135.9, 140.3, 145.8. IR (ATR) cm−1: 1608, 1553, 1473, 1451, 1368. Anal. Calcd for C17H19N3: C, 76.95; H, 7.22; N, 15.84. Found: C, 77.05; H, 7.28; N, 16.02.
N-Benzyl-1-ethyl-2,3-dimethyl-1H-pyrrolo[2,3-c]pyridine-7-amine Hydrochloride (7b)The obtained free base was dissolved in methanol (5 mL), 10% hydrogen chloride methanol solution (2 mL) was added, and the mixture was concentrated under reduced pressure. The residue was crystallized from ethanol to give the title compound as a colorless solid (24%). mp 232–235°C. 1H-NMR (DMSO-d6) δ: 1.24 (3H, t, J=7.4 Hz), 2.17 (3H, s), 2.42 (3H, s), 4.53 (2H, q, J=7.4 Hz), 4.89 (2H, d, J=6.2 Hz), 7.10 (1H, d, J=7.0 Hz), 7.28–7.46 (6H, m), 8.00–8.20 (1H, m), 12.62 (1H, br s). 13C-NMR (DMSO-d6) δ: 8.3, 10.2, 17.0, 40.2, 44.8, 105.3, 109.6, 116.1, 124.5, 127.0, 127.3, 128.5, 134.2, 137.3, 141.0, 142.5. IR (ATR) cm−1: 1643, 1603. ESI-HR-MS m/z: 280.1811 (Calcd for C18H21N3: 280.1808 [M+H]+).
1-Ethyl-N-(4-fluoro-2-methylbenzyl)-2,3-dimethyl-1H-pyrrolo[2,3-c]pyridine-7-amine (7c): Colorless solid (75%). mp 102–104°C. 1H-NMR (CDCl3) δ: 1.29 (3H, t, J=7.2 Hz), 2.18 (3H, s), 2.30 (3H, s), 2.41 (3H, s), 4.16 (2H, q, J=7.2 Hz), 4.35 (1H, br t, J=5.1 Hz), 4.68 (2H, d, J=5.1 Hz), 6.82–6.90 (3H, m), 7.29–7.34 (1H, m), 7.78 (1H, d, J=5.4 Hz). 13C-NMR (CDCl3) δ: 8.9, 10.0, 17.4, 19.2, 40.1, 43.7, 105.5, 107.6, 112.5, 112.7, 116.9, 117.2, 120.2, 130.0, 130.2, 133.65, 133.69, 133.9, 134.5, 135.9, 139.0, 139.1, 145.1, 160.4, 163.6. IR (ATR) cm−1: 1608, 1555, 1496, 1474, 1367. Anal. Calcd for C19H22FN3: C, 73.28; H, 7.12; N, 13.49. Found: C, 73.06; H, 7.29; N, 13.48.
N-(4-Fluoro-2-methylbenzyl)-2,3-dimethyl-1-propyl-1H-pyrrolo[2,3-c]pyridine-7-amine (7d): Colorless solid (33%). mp 88–89°C. 1H-NMR (CDCl3) δ: 0.78 (3H, t, J=7.8 Hz), 1.66–1.74 (2H, m), 2.17 (3H, s), 2.29 (3H, s), 2.41 (3H, s), 3.99–4.04 (2H, m), 4.28 (1H, br t, J=5.1 Hz), 4.65 (2H, d, J=5.1 Hz), 6.85–6.94 (3H, m), 7.29–7.34 (1H, m), 7.78 (1H, d, J=5.4 Hz). 13C-NMR (CDCl3) δ: 8.9, 10.3, 10.8, 19.1, 25.9, 43.8, 47.0, 105.5, 107.3, 112.5, 112.8, 116.9, 117.2, 120.4, 130.2, 130.3, 133.66, 133.70, 133.8, 134.9, 135.9, 139.1, 139.2, 145.2, 160.4, 163.7. IR (ATR) cm−1: 1607, 1554, 1496, 1471, 1367. Anal. Calcd for C20H24FN3: C, 73.82; H, 7.43; N, 12.91. Found: C, 73.76; H, 7.42; N, 12.55.
N-(4-Fluoro-2-methylbenzyl)-1-isobutyl-2,3-dimethyl-1H-pyrrolo[2,3-c]pyridine-7-amine (7e): Colorless solid (65%). mp 102–103°C. 1H-NMR (CDCl3) δ: 0.74 (6H, d, J=6.9 Hz), 2.01–2.10 (1H, m), 2.17 (3H, s), 2.27 (3H, s), 2.41 (3H, s), 3.83 (2H, d, J=7.5 Hz), 4.24 (1H, br t, J=4.5 Hz), 4.64 (2H, d, J=4.5 Hz), 6.83–6.94 (3H, m), 7.29–7.34 (1H, m), 7.78 (1H, d, J=5.7 Hz). 13C-NMR (CDCl3) δ: 9.0, 10.9, 19.1, 19.6, 32.0, 43.9, 52.8, 105.4, 107.2, 112.5, 112.8, 116.9, 117.2, 120.6, 130.4, 130.5, 133.6, 133.7, 134.0, 135.4, 136.0, 139.2, 139.3, 145.3, 160.5, 163.7. IR (ATR) cm−1: 1606, 1554, 1496, 1469, 1369. Anal. Calcd for C21H26FN3: C, 74.30; H, 7.72; N, 12.38. Found: C, 74.13; H, 7.65; N, 12.19.
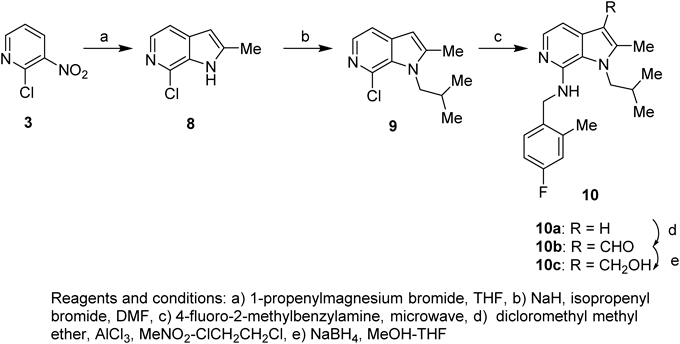
The following compounds 8, 9, and 10a were prepared in a similar manner with compounds 4, 6, and 7, respectively.
7-Chloro-2-methyl-1H-pyrrolo[2,3-c]pyridine (8): A pale-yellow solid (28%). mp 160–161°C. 1H-NMR (CDCl3) δ: 2.52 (3H, s), 6.30 (1H, s), 7.34 (1H, d, J=5.6 Hz), 7.98 (1H, d, J=5.6 Hz), 8.46 (1H, br).
7-Chloro-1-isobutyl-2-methyl-1H-pyrrolo[2,3-c]pyridine (9): An oil (81%). 1H-NMR (CDCl3) δ: 0.90 (6H, d, J=6.6 Hz), 2.21–2.30 (1H, m), 2.45 (3H, d, J=0.9 Hz), 4.23 (2H, br d, J=6.9 Hz), 6.29 (1H, d, J=0.9 Hz), 7.31 (1H, d, J=5.4 Hz), 7.92 (1H, d, J=5.4 Hz).
N-(4-Fluoro-2-methylbenzyl)-1-isobutyl-2-methyl-1H-pyrrolo[2,3-c]pyridine-7-amine (10a): A colorless solid (88%). mp 132–133°C. 1H-NMR (CDCl3) δ: 0.76 (6H, d, J=6.9 Hz), 2.04–2.18 (1H, m), 2.36 (3H, d, J=0.9 Hz), 2.42 (3H, s), 3.84 (2H, d, J=7.5 Hz), 4.25 (1H, br t, J=4.8 Hz), 4.65 (2H, d, J=4.8 Hz), 6.16 (1H, d, J=0.9 Hz), 6.85–6.96 (3H, m), 7.31–7.35 (1H, m), 7.78 (1H, d, J=5.4 Hz).
7-[(4-Fluoro-2-methylbenzyl)amino]-1-isobutyl-2-methyl-1H-pyrrolo[2,3-c]pyridine-3-carbaldehyde (10b)10a (339 mg, 1.0 mmol) was dissolved in nitromethane (4 mL) and 1,2-dichloroethane (4 mL), and aluminum(III) chloride (145 mg, 1.1 mmol) and dichloromethyl methyl ether (0.1 mL, 1.1 mmol) were added at 0°C. After stirring at the same temperature for 30 min, the same amount of aluminum(III) chloride and dichloromethyl methyl ether was added twice, and the mixture was stirred for 1 h. The reaction mixture was basified with 8 mol/L aqueous sodium hydroxide solution, and the mixture was extracted with EtOAc. The extract was washed with saturated brine, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (eluent: hexane–EtOAc=1 : 1) to give the title compound as a colorless solid (259 mg, 0.73 mmol, 73%). mp 129–130°C. 1H-NMR (CDCl3) δ: 0.81 (6H, d, J=6.6 Hz), 2.11–2.20 (1H, m), 2.41 (3H, s), 2.66 (3H, s), 3.90 (2H, d, J=6.9 Hz), 4.26 (1H, br t, J=4.5 Hz), 4.65 (2H, d, J=4.5 Hz), 6.86–6.97 (2H, m), 7.30–7.34 (1H, m), 7.57 (1H, d, J=5.7 Hz), 7.98 (1H, d, J=5.7 Hz), 10.18 (1H, s).
{7-[(4-Fluoro-2-methylbenzyl)amino]-1-isobutyl-2-methyl-1H-pyrrolo[2,3-c]pyridin-3-yl}methanol (10c)10b (216 mg, 0.61 mmol) was dissolved in methanol (5 mL) and THF (5 mL), and sodium borohydride (41 mg, 1.1 mmol) was added at 0°C. After stirring at room temperature for 1 h, several drops of acetic acid were added for a treatment, and the solvent was evaporated under reduced pressure. A 6% aqueous sodium hydrogen carbonate solution was added to the residue and the mixture was extracted with EtOAc. The extract was washed with saturated brine, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The residue was recrystallized from EtOAc–hexane to give the title compound as a yellow powder (145 mg, 0.41 mmol, 67%). mp 156–157°C. 1H-NMR (CDCl3) δ: 0.76 (6H, d, J=6.3 Hz), 1.25 (1H, t, J=5.1 Hz), 2.03–2.14 (1H, m), 2.39 (3H, s), 2.41 (3H, s), 3.86 (2H, d, J=6.9 Hz), 4.28 (1H, br t, J=4.8 Hz), 4.65 (2H, d, J=4.8 Hz), 4.77 (2H, d, J=5.1 Hz), 6.85–6.96 (2H, m), 7.01 (1H, d, J=5.4 Hz), 7.30–7.35 (1H, m), 7.84 (1H, d, J=5.4 Hz). 13C-NMR (CDCl3) δ: 10.9, 19.1, 19.6, 31.9, 43.8, 52.8, 55.8, 105.0, 111.8, 112.6, 112.9, 117.0, 117.3, 120.7, 130.5, 130.6, 133.1, 133.40, 133.44, 136.9, 137.0, 139.2, 139.3, 145.4, 160.5, 163.8. IR (ATR) cm−1: 1604, 1554, 1497, 1472, 1373. Anal. Calcd for C21H26FN3O: C, 70.96; H, 7.37; N, 11.82. Found: C, 70.72; H, 7.31; N, 11.68.
Proton Potassium—Adenosine Triphosphatase (H+/K+-ATPase) Inhibitory Activity TestAccording to the method of Wallmark et al.,15) a gastric mucosal membrane microsomal fraction was prepared from the stomach of swine. First, the stomach was removed, washed with tap water, immersed in 3 mol/L brine, and the surface of the mucosal membrane was wiped with a paper towel. The gastric mucosal membrane was detached, chopped, and homogenized in a 0.25 mol/L saccharose solution (pH 6.8) containing 1 mmol/L ethylenediaminetetraacetic acid (EDTA) and 10 mmol/L Tris-hydrochloric acid using polytron (Kinematica). The obtained homogenate was centrifuged at 20000×g for 30 min and the supernatant was centrifuged at 100000×g for 90 min. The precipitate was suspended in 0.25 mol/L saccharose solution, superimposed on a 0.25 mol/L saccharose solution containing 7.5% Ficoll, and centrifuged at 100000×g for 5 h. The fraction containing the interface between the both layers was recovered, and centrifugally washed with 0.25 mol/L saccharose solution. The obtained microsomal fraction was used as a proton, potassium–adenosine triphosphatase standard product. To 40 mL of a 50 mmol/L N-(2-hydroxyethyl)piperazine-N′-2-ethanesulfonic acid (HEPES)-Tris buffer (5 mmol/L magnesium chloride, 10 mmol/L potassium chloride, 10 µmol/L valinomycin, pH=6.5) containing 2.5 µg/mL (based on the protein concentration) of the enzyme standard product was added a test compound (5 mL) dissolved in a 10% aqueous dimethyl sulfoxide solution, and the mixture was incubated at 37°C for 30 min. The enzyme reaction was started by adding 5 mL of a 2 mmol/L adenosine triphosphate tris salt solution (50 mmol/L HEPES-Tris buffer (5 mmol/L magnesium chloride, pH 6.5)). The enzyme reaction was carried out at 37°C for 20 min, and 15 mL of a malachite green solution (0.12% malachite green solution in sulfuric acid (2.5 mol/L), 7.5% ammonium molybdate and 11% Tween 20 were mixed at a ratio of 100 : 25 : 2) was added to quench the reaction. After allowing to stand at room temperature for 15 min, the resulting reaction product of inorganic phosphorus with malachite green was colorimetrically determined at a wavelength of 610 nm. In addition, the amount of the inorganic phosphoric acid in the reaction solution free of potassium chloride was measured in the same manner, which was subtracted from the inorganic phosphoric acid amount in the presence of potassium chloride to determine the H+/K+-ATPase activity. The inhibitory rate (%) was determined from the activity value of the control and the activity values of various concentrations of the test compound, and the 50% inhibitory concentration (IC50) of the H+/K+-ATPase activity was determined.
Inhibiton of Histamine-Stimulated Acid Secretion in Anesthetized Rats (i.v.)Animal experiments were carried out in accordance with ethical guidelines established by the Animal Care and Use Committee at Takeda Pharmaceutical Company, Ltd. Seven-week-old male Jcl: Sprague-Dawley (SD) rats were used. The animals were fasted for 24 h but had free access to water before the experiment. The pylorus was ligated after anesthetization with urethane (1.2 g/kg, intraperitoneally (i.p.)) and the abdomen was closed. Drugs and the vehicle were given intravenously just after the pylorus ligation. Three minutes later, histamine 2HCl (30 mg/kg/10 mL) was injected subcutaneously. Three hours after histamine administration, the rats were sacrificed by CO2 asphyxiation and the stomachs were removed. The gastric contents were collected and centrifuged at 3000 rpm for 10 min. The volume of each sample was measured and the acid concentration was determined by automatic titration to pH 7.0 with 0.1 mol/L NaOH (COM-555SC; Hiranuma Sangyo Co., Ltd., Japan), and the total acid output during the 3 h period (mEq/3 h) was calculated.