Experimental
General RemarksMaterials were obtained from commercial suppliers and used without further purification unless otherwise mentioned. All reactions were carried out in oven-dried glassware under a slight positive pressure of argon unless otherwise noted. Anhydrous THF and CH2Cl2 were purchased from Kanto Chemical Co., Inc. Anhydrous toluene, DMF, and MeCN were purchased from Wako Pure Chemical Industries, Ltd. Anhydrous EtOAc, MeOH, 1,2-dichloroethane, and EtOH were dried and distilled according to the standard protocols. Column chromatography was performed on Silica Gel 60N (Kanto, spherical neutral, 63–210 µm) and flash column chromatography was performed on Silica Gel 60N (Kanto, spherical neutral, 40–50 µm) using the indicated eluent. Preparative TLC and analytical TLC were performed on Merck 60 F254 glass plates precoated with a 0.25 mm thickness of silica gel. All melting points were determined on a Yanaco micro melting point apparatus and uncorrected. IR spectra were measured on a SHIMADZU FTIR-8300 spectrometer or a JASCO FT/IR-4100 spectrometer. NMR spectra were recorded on a JEOL-ECA600, a GX500 spectrometer and JNM-AL400 spectrometer with tetramethylsilane (0 ppm) or chloroform (7.26 ppm) as an internal standard. Mass spectra were recorded on a JEOL JMS-DX-303 spectrometer, a JMS-AX-500 spectrometer, a JMS-T100GC spectrometer, or a MS-50070BU spectrometer. Optical rotations were measured on a Horiba SEPA-300 high sensitive polarimeter. Elemental analyses were performed by a Yanaco CHN CORDER MT-5.
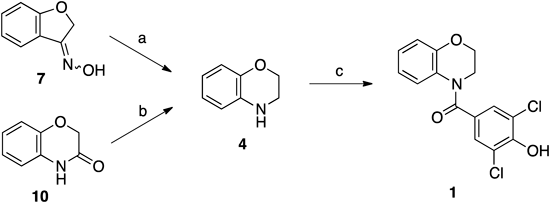
3,5-Dichloro-4-hydroxybenzoyl ChlorideA screw top test tube equipped with a magnetic stirring bar was charged with 3,5-dichloro-4-hydroxybenzoic acid (57.8 mg, 0.279 mmol) and anhydrous dichloromethane (0.56 mL). To the solution were added oxalyl chloride (36 µL, 0.42 mmol) and N,N-dimethylformamide (DMF) (2.0 µL, 0.026 mmol) at 0°C. After the reaction mixture was stirred at 0°C for 5 min, the mixture was allowed to warm to room temperature and the solution was stirred for 1.5 h. The mixture was concentrated under reduced pressure to afford 3,5-dichloro-4-hydroxybenzoyl chloride as a yellow solid, which was used to the next reaction without further purification.
2,6-Dichloro-4-(2,3-dihydro-1,4-benzoxazin-4-ylcarbonyl)phenol (1)A screw top test tube equipped with a magnetic stirring bar was charged with benzoxazine 4 (18.8 mg, 0.139 mmol) and anhydrous EtOAc (0.30 mL). To the stirred solution was added the crude acid chloride (0.279 mmol) in anhydrous EtOAc (0.26 mL) at room temperature. Stirring was continued for 4 h and the reaction mixture was quenched with saturated aqueous NaHCO3 and extracted with EtOAc three times. The combined organic extracts were washed with brine, dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure to leave the residue, which was purified by preparative TLC (hexanes–EtOAc=3 : 1) to afford the URAT-1 inhibitor 1 (12.9 mg, 0.0398 mmol, 29%) as a colorless solid; Rf=0.21 (Silica gel, hexanes–EtOAc=3 : 1); IR (neat) cm−1: 2927, 1618, 1597, 1489, 1399, 1240, 1179, 1059, 755; 1H-NMR (400 MHz, acetone-d6) δ: 7.55 (2H, s), 7.25–7.11 (1H, m), 7.00 (1H, ddd, J=8.4, 7.6, 1.6 Hz), 6.88 (1H, dd, J=8.4, 1.6 Hz), 6.73 (1H, ddd, J=8.4, 7.6, 1.6 Hz), 4.43–4.33 (2H, m), 4.02–3.91 (2H, m), 2.81 (1H, br s); 13C-NMR (100 MHz, acetone-d6) δ: 166.5, 151.8, 147.3, 129.9, 129.6, 127.0, 126.1, 125.1, 122.4, 120.4, 117.9, 66.8, 44.2; high resolution (HR)-MS (electrospray ionization (ESI)) Calcd for C15H12Cl2NO3 (M+H+): 324.0189; Found: 324.0173.
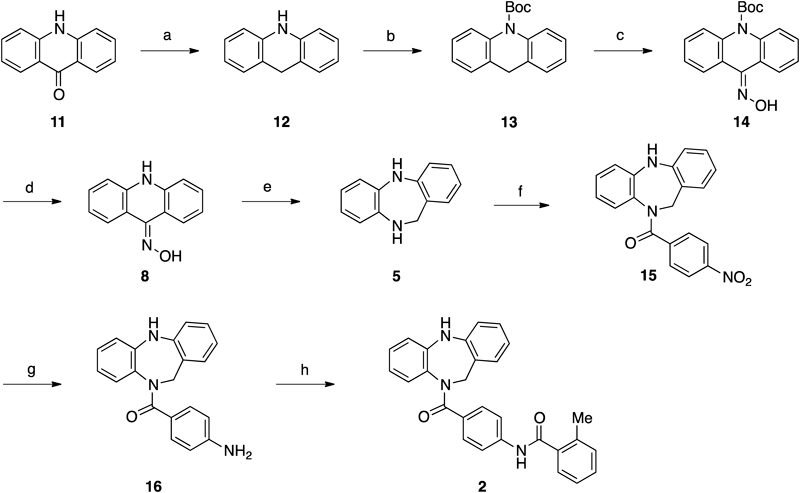
tert-Butyl Acridine-10(9H)-carboxylate (13)A two-necked 20-mL round-bottomed flask equipped with a magnetic stirring bar was charged with acridone 11 (195 mg, 1.00 mmol), and THF (2.5 mL). To the solution was added BH3·THF (1.08 M in THF, 1.3 mL, 1.4 mmol) at room temperature. The reaction mixture was heated at reflux for 4 h. The reaction was quenched with brine and 2 M aqueous NaOH, and the mixture was extracted with ether three times. The combined organic extracts were washed with saturated aqueous NaHCO3, dried over anhydrous magnesium sulfate, and filtered. The filtrate was concentrated under reduced pressure to afford the crude dihydroacridine 12 as a colorless solid, which was used to the next reaction without further purification. A 50-mL round-bottomed flask equipped with a magnetic stirring bar was charged with 12, Boc2O (766 mg, 3.51 mmol), and MeCN (3.3 mL). To the solution was added DMAP (203 mg, 1.66 mmol) at 0°C and stirring was continued for 11 h at room temperature. The reaction was diluted with ether. The mixture was washed with saturated aqueous NH4Cl and brine, dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure to leave the residue, which was purified by silica gel column chromatography (hexanes–EtOAc=9 : 1) to afford 13 (226 mg, 0.803 mmol, 80%); mp 97.4–98.9°C, colorless crystals; Rf=0.47 (Silica gel, hexanes–EtOAc=9 : 1); IR (neat) cm−1: 1706, 1476, 1328, 1269, 1253, 1151, 760; 1H-NMR (400 MHz, CDCl3) δ: 7.63 (d, 2H, J=8.0 Hz), 7.36–7.17 (m, 4H), 7.12 (d, 2H, J=7.2 Hz), 3.80 (s, 2H), 1.54 (s, 9H); 13C-NMR (125 MHz, CDCl3) δ: 152.5, 138.8, 133.0, 126.9, 125.9, 125.04, 124.96, 33.8, 28.2, 27.9; HR-MS (ESI) Calcd for C14H12NO2 [(M−t-Bu+2H)+] 226.0863, Found: 226.0878.
tert-Butyl 9-(Hydroxyimino)acridine-10(9H)-carboxylate (14)A two-necked 30-mL round-bottomed flask equipped with a magnetic stirring bar was charged with tert-butyl acridine-10(9H)-carboxylate 13 (100.5 mg, 0.36 mmol) and anhydrous THF (3.6 mL). To the solution was added t-BuONO (51.0 µL, 0.43 mmol) at 0°C, followed by KHMDS (0.47 M in toluene, 0.91 mL, 0.43 mmol) dropwise at 0°C. After 0.5 h, the reaction was quenched with saturated aqueous NH4Cl, and the aqueous layer was extracted three times with ether. The combined organic extracts were washed with brine, dried over anhydrous sodium sulfate, and filtered. The organic solvents were removed under reduced pressure to leave the residue, which was purified by flash silica gel column chromatography (hexanes–EtOAc=4 : 1) to afford oxime 14 (103.6 mg, 0.33 mmol, 93%) as a colorless solid; Rf=0.35 (hexanes–EtOAc=4 : 1); IR (neat) cm−1: 3071, 2974, 2928, 1716, 1598, 1463, 1322, 1251, 1159, 955, 923, 755; 1H-NMR (400 MHz, CDCl3) δ: 8.42 (dd, 1H, J=8.0, 1.2 Hz), 8.23 (br s, 1H), 7.79 (d, 1H, J=8.0 Hz), 7.78 (d, 1H, J=8.0 Hz), 7.72 (dd, 1H, J=8.0, 1.2 Hz), 7.41 (ddd, 1H, J=8.0, 8.0, 1.2 Hz), 7.39 (ddd, 1H, J=8.0, 8.0, 1.2 Hz), 7.27 (dd, 1H, J=8.0, 8.0 Hz), 7.24 (dd, 1H, J=8.0, 8.0 Hz), 1.53 (s, 9H); 13C-NMR (100 MHz, CDCl3) δ: 152.0, 146.4, 138.5, 137.5, 129.2, 129.0, 128.3, 128.2, 125.47, 125.42, 124.7, 124.6, 124.3, 124.2, 82.9, 28.1; HR-MS (ESI) Calcd for C18H19N2O3 (M+H+) 311.1390, Found: 311.1386; Anal. Calcd for C18H18N2O3: C, 69.66; H, 5.85; N, 9.03; Found: C, 69.79; H, 5.81; N, 8.94.
9(10H)-Acridone Oxime (8)A 20-mL round-bottomed flask equipped with a magnetic stirring bar was charged with carbamate 14 (32.6 mg, 0.105 mmol) and anhydrous dichloromethane (0.7 mL). To the solution was added trifluoroacetic acid (TFA) (0.35 mL, 4.7 mmol) at 0°C. After the reaction mixture was stirred at 0°C for 5 min, the mixture was allowed to warm to room temperature and the solution was stirred for 3 h. The reaction mixture was quenched with saturated aqueous NaHCO3 at 0°C and was extracted with EtOAc three times. The combined organic extracts were washed with brine, dried over anhydrous sodium sulfate, and filtered. The filtrate was concentrated under reduced pressure to afford the crude acridone oxime 8, which was subjected to the next reaction without further purification.
(5H-Dibenzo[b,e][1,4]diazepin-10(11H)-yl)(4-nitrophenyl)methanone (15)A 20-mL round-bottomed flask equipped with a magnetic stirring bar was charged with oxime 8 and anhydrous dichloromethane (1.0 mL). To the stirred solution was added DIBALH (1.03 M in hexane, 0.60 mL, 0.62 mmol) at 0°C. After the reaction mixture was stirred at 0°C for 5 min, the mixture was allowed to warm to room temperature and the solution was stirred for 2 h. The reaction was quenched with NaF (127 mg) and H2O (40 µL) at 0°C, and the resulting mixture was stirred for 30 min at the same temperature. Then the mixture was filtered through a pad of Celite. The filter cake was washed with ether and dichloromethane, and the filtrate was concentrated under reduced pressure to afford the crude dibenzodiazepine 5 as yellow solids, which was subjected to the next reaction without further purification. A 20-mL round-bottomed flask equipped with a magnetic stirring bar was charged with 5, Et3N (73 µL, 0.52 mmol), and anhydrous dichloromethane (1.0 mL). To the solution was added p-nitrobenzoyl chloride (39.3 mg, 0.212 mmol) at 0°C. After the reaction mixture was stirred at 0°C for 5 min, the mixture was allowed to warm to room temperature and the solution was stirred for 1 h. The reaction was quenched with saturated aqueous NH4Cl, and the mixture was extracted with dichloromethane three times. The combined organic extracts were washed with brine, dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure to leave the residue, which was purified by silica gel column chromatography (hexanes–EtOAc=3 : 1 to 3 : 2) to afford amide 15 (29.1 mg, 0.0843 mmol, 80% for 3 steps) as a yellow solid; Rf=0.63 (Silica gel, hexanes–EtOAc=1 : 1); IR (neat) cm−1: 3337, 1634, 1595, 1520, 1504, 1495, 1344, 745, 731; 1H-NMR (400 MHz, CDCl3) δ: 8.00 (d, 2H, J=8.4 Hz), 7.41 (d, 2H, J=8.4 Hz), 7.29 (d, 1H, J=8.0 Hz), 7.19 (dd, 1H, J=8.0, 8.0 Hz), 7.13–7.00 (m, 1H), 6.95–6.80 (m, 3H), 6.62–6.50 (m, 2H), 6.43 (br s, 1H), 5.81 (br s, 1H), 4.27 (br s, 1H); 13C-NMR (100 MHz, CDCl3) δ: 167.0, 148.1, 142.1, 140.9, 138.6, 130.6, 130.2, 129.2, 128.8, 128.7, 128.3, 123.5, 123.0, 119.9, 119.8, 118.5, 117.6, 51.9; HR-MS (EI) Calcd for C20H15N3O3 (M+) 345.1113, Found: 345.1087.
N-(4-(10,11-Dihydro-5H-dibenzo[b,e][1,4]diazepine-10-carbonyl)phenyl)-2-methylbenzamide (2)A Schlenk tube equipped with a magnetic stirring bar was charged with amide 15 (13.1 mg, 0.038 mmol), 10% Pd/C (10.6 mg, 0.010 mmol) and EtOAc (0.38 mL). To the flask was charged with hydrogen gas (1 atm). The resulting suspension was vigorously stirred for 2 h. The reaction mixture was filtered through a pad of Celite. The filter cake was washed with EtOAc, and the filtrate was concentrated under reduced pressure to leave the crude amine 16 as yellow solids, which was subjected to the next reaction without further purification. A 20-mL round-bottomed flask equipped with a magnetic stirring bar was charged with 16, Et3N (26 µL, 0.19 mmol), and anhydrous dichloromethane (0.76 mL). To the solution was added 2-methylbenzoyl chloride (7.5 µL, 0.057 mmol) at 0°C. After the reaction mixture was stirred at 0°C for 5 min, the mixture was allowed to warm to room temperature and the solution was stirred for 1 h. The reaction was quenched with saturated aqueous NH4Cl, and the mixture was extracted with dichloromethane three times. The combined organic extracts were washed with saturated aqueous NaHCO3 and brine, dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure to leave the residue, which was purified by preparative TLC (hexanes–EtOAc=1 : 1) to afford amide 2 (14.0 mg, 0.0323 mmol, 85% for 2 steps) as a yellow solid; Rf=0.39 (Silica gel, hexanes–EtOAc=1 : 1); IR (neat) cm−1: 3320, 3229, 3056, 3012, 1660, 1623, 1594, 1525, 1493, 1407, 1317, 752; 1H-NMR (400 MHz, CDCl3) δ: 7.92 (br s, 1H), 7.60–7.06 (m, 10H), 6.98 (dd, 1H, J=7.6, 7.6 Hz), 6.83 (d, 2H, J=8.0 Hz), 6.79 (dd, 1H, J=7.6, 7.6 Hz), 6.70 (br s, 1H), 6.63–6.45 (m, 2H), 5.80 (br s, 1H), 4.20 (br s, 1H), 2.36 (s, 3H); 13C-NMR (125 MHz, CDCl3) δ: 168.4, 168.1, 141.4, 139.5, 138.5, 136.4, 136.0, 132.0, 131.2, 130.3, 130.1, 129.7, 128.9, 128.5, 127.7, 126.9, 126.6, 125.8, 125.7, 123.8, 119.7, 119.4, 118.4, 117.4, 20.0, 19.7; HR-MS (ESI) Calcd for C28H24N3O2 (M+H+) 434.1863, Found: 434.1870.
Methyl 5-Bromo-2-ethynylbenzoate (18)A two-necked 200-mL round-bottomed flask equipped with a magnetic stirring bar was charged with aryl iodide 17 (1.733 g, 5.083 mmol), CuI (20.8 mg, 0.109 mmol), and DMF (25 mL). To the solution were added Et3N (2.1 mL, 15.1 mmol), PdCl2(PPh3)2 (36.1 mg, 0.0514 mmol), and trimethylsilylacetylene (1.1 mL, 7.8 mmol). The solution was stirred for 30 h. The reaction was quenched with water, and the mixture was extracted with EtOAc three times. The combined organic extracts were washed with water and brine, dried over anhydrous magnesium sulfate, filtered, and concentrated under reduced pressure to leave the residue, which was purified by short silica gel column chromatography (hexanes–EtOAc=9 : 1) to afford TMS-acetylene (1.706 g, 5.49 mmol, quant.) as yellow solids; Rf=0.74 (Silica gel, hexanes–EtOAc=3 : 1). A 100-mL round-bottomed flask equipped with a magnetic stirring bar was charged with TMS-acetylene and MeOH (10 mL). To the solution was added K2CO3 (1.4 g, 10.1 mmol) at 0°C. The solution was stirred for 10 min. The solution was concentrated under reduced pressure. The reaction mixture was filtered through a pad of Celite. The filter cake was washed with ether, and the filtrate was concentrated under reduced pressure to leave the residue, which was purified by silica gel column chromatography (hexanes–ether=9 : 1) to afford aryl acetylene 18 (909 mg, 3.80 mmol, 75% for 2 steps) as a pale yellow solid; Rf=0.50 (Silica gel, hexanes–ether=3 : 1); IR (neat) cm−1: 3256, 3099, 2950, 2103, 1730, 1430, 1291, 1243, 1073, 689; 1H-NMR (400 MHz, CDCl3) δ: 8.09 (d, 1H, J=2.0 Hz), 7.61 (dd, 1H, J=8.0, 2.0 Hz), 7.48 (d, 1H, J=8.0 Hz), 3.94 (s, 3H), 3.45 (s, 1H); 13C-NMR (100 MHz, CDCl3) δ: 165.1, 136.2, 134.8, 133.9, 133.3, 122.6, 121.6, 83.4, 81.1, 52.5; HR-MS (ESI) Calcd for C10H8BrO2 (M+H+) 238.9702, Found: 238.9697.
Procedure for the Sandmeyer Reaction to Give 3-Iodo-1-methylthiobenzeneA 300-mL round-bottomed flask equipped with a magnetic stirring bar was charged with m-(methylthio)aniline (4.00 mL, 32.5 mmol), crushed ice (24 g), and MeCN (24 mL). To the stirred solution was added concentrated H2SO4 (24 mL) at 0°C over 30 min. To the slurry was added aqueous NaNO2 (4.04 g, 58.6 mmol) in cold H2O (8 mL) at 0°C dropwise over 30 min to maintain the internal temperature below 5°C. After the mixture was stirred at 0°C for 30 min, the mixture was poured into a solution of KI (18.9 g, 114 mmol) in H2O (24 mL) at 0°C. After the resulting mixture was stirred at 0°C for 5 min, the mixture was allowed to warm to room temperature and the solution was stirred for 1 h. The reaction was quenched with H2O, and the mixture was extracted with CHCl3 three times. The combined organic extracts were washed with saturated aqueous NaHCO3, saturated aqueous Na2S2O3, and brine, dried over anhydrous sodium sulfate, and filtered. The filtrate was concentrated under reduced pressure to leave the residue, which was purified by silica gel column chromatography (hexanes) to afford 3-iodo-1-methylthiobenzene (7.71 g, 30.8 mmol, 95%) as a pale yellow oil. The spectral data of 3-iodo-1-methylthiobenzene was identical with those reported in ref. 14.
Methyl 5-Bromo-2-[(3-methylthiophenyl)ethynyl]benzoate (19b)A two-necked 200-mL round-bottomed flask equipped with a magnetic stirring bar was charged with aryl acetylene 18 (718 mg, 3.00 mmol), 3-iodo-1-methylthiobenzene (906 mg, 3.62 mmol), and DMF (6.0 mL). To the solution were added PdCl2(PPh3)2 (21.5 mg, 0.0306 mmol), CuI (11.4 mg, 0.0599 mmol), and Et3N (1.3 mL, 9.3 mmol). The solution was stirred for 3 h. The reaction was quenched with water, and the mixture was extracted with EtOAc three times. The combined organic extracts were washed with water and brine, dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure to leave the residue, which was purified by silica gel column chromatography (hexanes–EtOAc=19 : 1) to afford diaryl acetylene 19b (999 mg, 2.77 mmol, 92%) as a pale yellow solid; Rf=0.25 (Silica gel, hexanes–EtOAc=19 : 1); IR (neat) cm−1: 2214, 1726, 1561, 1475, 1437, 1286, 1241, 1092, 1078, 964, 817, 780, 679; 1H-NMR (400 MHz, CDCl3) δ: 8.12 (d, 1H, J=2.0 Hz), 7.61 (dd, 1H, J=8.4, 2.0 Hz), 7.49 (d, 1H, J=8.4 Hz), 7.42 (br s, 1H), 7.33 (ddd, 1H, J=6.8, 1.6, 1.6 Hz), 7.26 (dd, 1H, J=8.0, 6.8 Hz), 7.23 (ddd, 1H, J=6.8, 1.6, 1.6 Hz), 3.96 (s, 3H), 2.50 (s, 3H); 13C-NMR (100 MHz, CDCl3) δ: 165.2, 139.0, 135.2, 134.7, 133.5, 133.2, 129.1, 128.7, 128.3, 126.9, 123.6, 122.5, 121.9, 95.0, 87.6, 52.4, 15.6; HR-MS (ESI) Calcd for C17H14BrO2S (M+H+) 360.9892, Found: 360.9890.
Methyl 5-Bromo-2-(Phenylethynyl)benzoate (19a)A pale yellow solid; IR (neat) cm−1: 1727, 1492, 1438, 1289, 1244, 1077, 968, 825, 758, 690; 1H-NMR (400 MHz, CDCl3) δ: 8.12 (d, 1H, J=2.4 Hz), 7.61 (dd, 1H, J=8.4, 2.4 Hz), 7.59–7.54 (m, 2H), 7.50 (d, 1H, J=8.4 Hz), 7.41–7.32 (m, 3H), 3.97 (s, 3H); 13C-NMR (100 MHz, CDCl3) δ: 165.3, 135.2, 134.8, 133.5, 133.2, 131.7, 128.8, 128.4, 123.0, 122.7, 121.8, 95.5, 87.3, 52.4; HR-MS (ESI) Calcd for C16H12BrO2 (M+H+) 315.0015, Found: 315.0014.
Methyl 5-Bromo-2-phenethylbenzoate (20a)A two-necked 10-mL round-bottomed flask equipped with a magnetic stirring bar was charged with acetylene 19a (58.1 mg, 0.184 mmol), PtO2 (3.9 mg, 0.017 mmol), EtOAc (0.7 mL), EtOH (0.7 mL), and AcOH (0.4 mL). To the flask was charged with hydrogen gas (1 atm). The resulting suspension was vigorously stirred for 1.5 h. After the reaction mixture was filtered through a pad of Celite, the filter cake was washed with EtOAc. The filtrate was concentrated under reduced pressure to leave the residue, which was purified by silica gel column chromatography (hexanes–EtOAc=19 : 1) to afford ester 20a (52.2 mg, 0.164 mmol, 89%) as a yellow solid; Rf=0.35 (Silica gel, hexanes–EtOAc=19 : 1); IR (neat) cm−1: 1726, 1434, 1288, 1246, 1081, 967, 700; 1H-NMR (400 MHz, CDCl3) δ: 8.03 (d, 1H, J=2.4 Hz), 7.50 (dd, 1H, J=8.4 and 2.4 Hz), 7.33–7.24 (m, 2H), 7.24–7.16 (m, 3H), 7.05 (d, 1H, J=8.4 Hz), 3.90 (s, 3H), 3.28–3.16 (m, 2H), 2.93–2.83 (m, 2H); 13C-NMR (100 MHz, CDCl3) δ: 166.6, 142.6, 141.5, 134.8, 133.5, 132.9, 131.1, 128.5, 128.3, 126.0, 119.5, 52.2, 37.8, 36.2; HR-MS (ESI) Calcd for C15H12BrO [(M−OMe)+] 287.0066, Found: 287.0063.
Methyl 5-Bromo-2-(3-(Methylthio)phenethyl)benzoate (20b)A Parr pressure bomb equipped with a magnetic stirring bar was charged with acetylene 19b (917 mg, 2.54 mmol), PtO2 (28.9 mg, 0.127 mmol), and THF (12.7 mL). To the flask was charged with hydrogen gas (70 atm). The resulting suspension was vigorously stirred for 14 h. After the reaction mixture was filtered through a pad of Celite, the filter cake was washed with EtOAc. The filtrate was concentrated under reduced pressure to leave the residue, which was purified by silica gel column chromatography (hexanes–EtOAc=19 : 1) to afford ester 20b as a mixture of inseparable compounds (855 mg, <2.34 mmol, <92%) as a yellow solid; Rf=0.25 (Silica gel, hexanes–EtOAc=19 : 1); IR (neat) cm−1: 2949, 2921, 1725, 1591, 1479, 1434, 1291, 1244, 1078, 967, 788; 1H-NMR (400 MHz, CDCl3) δ: 8.03 (d, 1H, J=2.4 Hz), 7.50 (dd, 1H, J=8.0, 2.4 Hz), 7.19 (dd, 1H, J=8.0, 8.0 Hz), 7.13–7.06 (m, 2H), 7.04 (d, 1H, J=8.0 Hz), 6.99–6.92 (m, 1H), 3.89 (s, 3H), 3.26–3.14 (m, 2H), 2.92–2.88 (m, 2H), 2.45 (s, 3H); 13C-NMR (100 MHz, CDCl3) δ: 166.4, 142.4, 142.1, 138.2, 134.8, 133.5, 132.8, 131.1, 128.8, 126.9, 125.4, 124.3, 119.6, 52.2, 37.7, 35.9, 15.8; HR-MS (ESI) Calcd for C16H14BrOS [(M–OMe)+] 332.9943, Found: 332.9915.
3-Bromo-10,11-dihydro-5H-dibenzo[a,d][7]annulen-5-one (22a)A 10-mL round-bottomed flask equipped with a magnetic stirring bar was charged with ester 20a (38.0 mg, 0.119 mmol), THF (0.6 mL), and MeOH (0.6 mL). To the solution was added 2 M aqueous NaOH (1.2 mL) at 0°C. After the reaction mixture was stirred at 0°C for 5 min, the mixture was allowed to warm to room temperature and the solution was stirred for 11 h. The reaction was quenched with 2 M aqueous HCl at 0°C, and the mixture was extracted with EtOAc three times. The combined organic extracts were washed with brine, dried over anhydrous sodium sulfate, and filtered. The filtrate was concentrated under reduced pressure to afford the crude carboxylic acid 21a as a colorless solid, which was subjected to the next reaction without further purification. A screw top test tube equipped with a magnetic stirring bar was charged with carboxylic acid 21a. To the tube was added the PPMA solution, which was prepared from P2O5 (161 mg, 1.13 mmol) and MsOH (1.0 mL) at 80°C for 1 h. After the mixture was stirred for 0.5 h at 80°C, the reaction mixture was poured into ice-water and extracted with CHCl3. The combined extracts were washed with brine and dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure to leave the residue, which was purified by preparative TLC (hexanes–EtOAc=17 : 3) to afford ketone 22a (23.3 mg, 0.0811 mmol, 68% for 2 steps) as a yellow oil; Rf=0.51 (Silica gel, hexanes–EtOAc=17 : 3); IR (neat) cm−1: 3060, 2921, 2856, 1652, 1597, 1584, 1474, 1289, 841, 751; 1H-NMR (400 MHz, CDCl3) δ: 8.14 (d, 1H, J=2.4 Hz), 7.98 (dd, 1H, J=8.0, 2.4 Hz), 7.53 (dd, 1H, J=8.0, 1.6 Hz), 7.45 (ddd, 1H, J=8.0, 8.0, 1.6 Hz), 7.33 (ddd, 1H, J=8.0, 8.0, 1.2 Hz), 7.22 (dd, 1H, J=8.0, 1.2 Hz), 7.11 (d, 1H, J=8.0 Hz), 3.24–3.10 (m, 4H); 13C-NMR (100 MHz, CDCl3) δ: 193.9, 141.8, 140.8, 140.1, 138.0, 135.0, 133.3, 132.7, 131.1, 130.7, 129.3, 126.8, 120.5, 34.7, 34.4; HR-MS (ESI) Calcd for C15H12BrO (M+H+), 287.0066; Found: 287.0059.
7-Bromo-2-(Methylthio)-10,11-dihydro-5H-dibenzo[a,d]-[7]annulen-5-one (22b)Yellow solid; IR (neat) cm−1: 2918, 1622, 1573, 1558, 1261, 1112, 774; 1H-NMR (400 MHz, CDCl3) δ: 8.15 (d, 1H, J=2.4 Hz), 8.03 (d, 1H, J=8.0 Hz), 7.52 (dd, 1H, J=8.0, 2.4 Hz), 7.16 (dd, 1H, J=8.0, 2.0 Hz), 7.10 (d, 1H, J=8.0 Hz), 7.03 (d, 1H, J=2.0 Hz), 3.19–3.09 (m, 4H), 2.52 (s, 3H); 13C-NMR (100 MHz, CDCl3) δ: 192.1, 145.5, 142.8, 140.6, 140.2, 134.9, 133.8, 133.5, 131.8, 130.8, 125.7, 123.4, 120.6, 35.3, 34.3, 14.7; HR-MS (ESI) Calcd for C16H14BrOS (M+H+) 332.9943, Found: 332.9921.
3-Bromo-10,11-dihydro-5H-dibenzo[a,d][7]annulen-5-one Oxime (9a)A 20-mL round-bottomed flask equipped with a magnetic stirring bar was charged with ketone 22a (608 mg, 2.12 mmol) and pyridine (4.2 mL). To the solution was added hydroxylamine hydrochloride (1.8 g, 26 mmol). The solution was stirred at 130°C for 48 h. The reaction was quenched with water. The resulting mixture was extracted with EtOAc three times. The combined organic extracts were washed with brine, dried over anhydrous sodium sulfate, and filtered. The filtrate was concentrated under reduced pressure to leave the residue, which was purified by silica gel column chromatography (hexanes–EtOAc=17 : 3) to afford oxime 9a (577 mg, 1.91 mmol, 90%) as a pale yellow solid; Rf=0.53 (Silica gel, hexanes–EtOAc=3 : 1); IR (neat) cm−1: 3172, 3059, 2916, 1426, 1311, 1006, 935, 819, 745; 1H-NMR (400 MHz, CDCl3) δ: 7.92 (br s, 0.55H), 7.82–7.70 (m, 0.9H), 7.62–7.57 (m, 0.55H), 7.57–7.51 (m, 0.55H), 7.48–7.09 (m, 5H), 7.05–6.97 (m, 0.45H), 3.24–2.96 (m, 4H); 13C-NMR (100 MHz, CDCl3) δ: 157.6, 157.3, 138.5, 138.1, 137.8, 137.4, 136.1, 135.4, 133.8, 133.0, 132.2, 132.0, 132.0, 131.1, 131.0, 130.5, 130.0, 129.6, 129.5, 128.6, 128.4, 128.3, 126.4, 125.8, 119.6, 119.3, 33.3, 33.1, 31.8, 31.5; HR-MS (ESI) Calcd for C15H13BrNO (M+H+) 302.0175, Found: 302.0176.
7-Bromo-2-methylthio-10,11-dihydro-5H-dibenzo[a,d][7]-annulen-5-one Oxime (9b)Pale yellow solid; IR (neat) cm−1: 3214, 1588, 1427, 1002, 931, 812; 1H-NMR (400 MHz, acetone-d6) δ: 10.64 (br s, 0.5H), 10.60 (br s, 0.5H), 7.70 (d, 0.5H, J=2.4 Hz), 7.56 (d, 0.5H, J=1.6 Hz), 7.54–6.80 (m, 5H), 3.20–2.95 (m, 4H), 2.49 (s, 1.5H), 2.46 (s, 1.5H); 13C-NMR (100 MHz, acetone-d6) δ: 140.7, 140.6, 140.0, 139.9, 138.84, 138.77, 138.2, 137.6, 133.3, 132.3, 132.14, 132.09, 132.06, 132.01, 131.95, 131.5, 131.3, 130.9, 130.3, 130.3, 128.3, 126.1, 124.4, 123.8, 119.6, 119.3, 34.1, 33.8, 32.4, 31.9, 15.1, 15.0; HR-MS (ESI) Calcd for C16H15BrNOS (M+H+) 348.0052, Found: 348.0037.
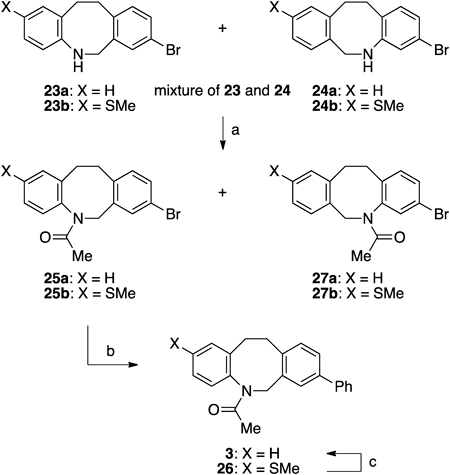
8-Bromo-5,6,11,12-tetrahydrodibenz[b,f]azocine (23a) and 3-Bromo-5,6,11,12-tetrahydrodibenz[b,f]azocine (24a)A screw top test tube equipped with a magnetic stirring bar was charged with oxime 9a (15.6 mg, 0.0516 mmol) and anhydrous 1,2-dichloroethane (0.26 mL). To the solution was added DIBALH (1.03 M in hexane, 0.30 mL, 0.31 mmol) dropwise at 80°C for 0.5 h. The solution was stirred for 15 h. The reaction mixture was cooled to 0°C, and diluted with ether. The reaction was quenched with methanol and 2 M aqueous NaOH, and the mixture was extracted with ether three times. The combined organic extracts were washed with brine, dried over anhydrous sodium sulfate, and filtered. The filtrate was concentrated under reduced pressure to leave the residue, which was purified by preparative TLC (hexanes–EtOAc=17 : 3) to afford a mixture of 23a and 24a (10.8 mg, 0.0375 mmol, 73%) as a yellow solid.
8-Bromo-2-methylthio-5,6,11,12-tetrahydrodibenz[b,f]azocine (23b) and 3-Bromo-9-methylthio-5,6,11,12-tetrahydrodibenz[b,f]azocine (24b)A two-necked 30-mL round-bottomed flask equipped with a magnetic stirring bar was charged with oxime 9b (175 mg, 0.503 mmol) and anhydrous dichloromethane (5.0 mL). To the stirred solution was added DIBALH (1.03 M in hexane, 3.0 mL, 3.09 mmol) dropwise at room temperature in water bath for 10 min. The solution was stirred for 12 h. The reaction mixture was cooled to 0°C and diluted with ether. The reaction was quenched with methanol and 2 M aqueous NaOH, and the mixture was extracted with diethyl ether three times. The combined organic extracts were washed with brine, dried over anhydrous sodium sulfate, and filtered. The filtrate was concentrated under reduced pressure to leave the residue, which was purified by silica gel column chromatography (hexanes–EtOAc=9 : 1) to afford an inseparable mixture of 23b and 24b (113 mg, 0.337 mmol, 67%) as a yellow solid; IR (neat) cm−1: 3376 (br), 2919, 2891, 1592, 1484, 1258, 813, 756; 1H-NMR (400 MHz, CDCl3, isomeric mixture (6 : 1)). Major isomer: δ: 7.22–7.15 (m, 2H), 6.96 (d, 1H, J=2.0 Hz), 6.90 (dd, 1H, J=8.0, 2.0 Hz), 6.89 (d, 1H, J=7.6 Hz), 6.46 (d, 1H, J=8.0 Hz), 4.35 (s, 2H), 3.27–3.18 (m, 2H), 3.16–3.08 (m, 2H), 2.37 (s, 3H). Minor isomer: δ: 7.02–7.15 (m, 3H), 6.81 (d, 1H, J=8.0 Hz), 6.77 (dd, 1H, J=7.6, 2.0 Hz), 6.61 (d, 1H, J=2.0 Hz), 4.37 (s, 2H), 3.27–3.18 (m, 2H), 3.16–3.08 (m, 2H), 2.41 (s, 3H); 13C-NMR (100 MHz, CDCl3) δ: 148.5, 145.3, 140.7, 139.3, 137.3, 133.7, 132.5, 132.2, 132.1, 131.9, 131.8, 131.2, 130.22, 130.19, 129.7, 128.3, 127.7, 127.6, 125.8, 124.6, 122.5, 121.2, 119.9, 119.8, 51.1, 50.8, 35.4, 34.8, 32.1, 31.6, 17.9, 15.6; HR-MS (ESI) Calcd for C16H17BrNS (M+H+), 334.0260; Found: 334.0249.
5-Acetyl-8-bromo-5,6,11,12-tetrahydrodibenz[b,f]azocine (25a)A screw top test tube equipped with a magnetic stirring bar was charged with mixture of 23a and 24a, Et3N (25 µL, 0.18 mmol) and anhydrous dichloromethane (0.36 mL). To the solution were added Ac2O (7.0 µL, 0.074 mmol) and DMAP (2.2 mg, 0.018 mmol) at room temperature. The solution was stirred for 0.5 h. The reaction mixture was quenched with saturated aqueous NH4Cl, and the mixture was extracted with dichloromethane three times. The combined organic extracts were washed with brine, dried over anhydrous sodium sulfate, and filtered. The filtrate was concentrated under reduced pressure to leave the residue, which was purified by preparative TLC (hexanes–EtOAc=3 : 1) to afford acetamide 25a (7.9 mg, 0.024 mmol, 66%) as colorless plates and acetamide 27a (2.3 mg, 0.0070 mmol, 19%) as colorless plates.
5-Acetyl-8-bromo-5,6,11,12-tetrahydrodibenz[b,f]azocine (25a)mp 146.5–148.3°C, colorless crystals; IR (neat) cm−1: 2944, 1651, 1496, 1386, 1282, 724; 1H-NMR (400 MHz, CDCl3) δ: 7.25–7.12 (m, 4H), 7.10–7.00 (m, 2H), 6.96–6.88 (m, 1H), 5.75 (d, 1H, J=14.8 Hz), 4.02 (d, 1H, J=14.8 Hz), 3.26–3.12 (m, 2H), 2.94–2.72 (m, 2H), 1.80 (s, 3H); 13C-NMR (100 MHz, CDCl3) δ: 170.2, 140.5, 139.3, 139.0, 137.3, 132.7, 131.2, 131.1, 130.8, 128.7, 128.5, 128.0, 119.7, 52.0, 34.6, 30.9, 22.7; HR-MS (ESI) Calcd for C17H17BrNO (M+H+) 330.0488, Found: 330.0489.
5-Acetyl-3-bromo-5,6,11,12-tetrahydrodibenz[b,f]azocine (27a)mp 140.7–142.2°C, colorless crystals; IR (neat) cm−1: 3019, 2927, 1660, 1486, 1385, 1295, 761; 1H-NMR (600 MHz, CDCl3) δ: 7.31–7.19 (m, 2H), 7.16–7.01 (m, 4H), 6.91 (d, 1H, J=7.8 Hz), 5.76 (d, 1H, J=15.0 Hz), 4.07 (d, 1H, J=15.0 Hz), 3.30–3.18 (m, 1H), 3.16–3.07 (m, 1H), 2.93–2.77 (m, 2H), 1.82 (s, 3H); 13C-NMR (100 MHz, CDCl3) δ: 169.8, 142.0, 139.9, 138.7, 134.5, 132.6, 131.6, 131.5, 130.2, 129.4, 128.0, 126.5, 120.2, 52.5, 34.4, 31.0, 22.9; HR-MS (ESI) Calcd for C17H17BrNO (M+H+) 330.0488, Found: 330.0484.
5-Acetyl-5,6,11,12-tetrahydro-8-phenyldibenz[b,f]azocine (3)A screw top test tube equipped with a magnetic stirring bar was charged with aryl bromide 25a (10.8 mg, 0.0287 mmol), phenylboronic acid (6.7 mg, 0.055 mmol), K2CO3 (7.9 mg, 0.057 mmol), Pd(PPh3)4 (2.2 mg, 1.9 µmol), THF (0.15 mL), and H2O (0.04 mL). The reaction mixture was heated to reflux for 6 h. The reaction mixture was diluted with EtOAc, washed with brine, dried over anhydrous sodium sulfate, and filtered. The filtrate was concentrated under reduced pressure to leave the residue, which was purified by preparative TLC (hexanes–acetone=3 : 1) to afford 3 (6.7 mg, 0.020 mmol, 70%) as a colorless oil; IR (neat) cm−1: 3027, 2930, 1658, 1494, 1389, 765; 1H-NMR (400 MHz, CDCl3) δ: 7.49 (d, 2H, J=7.2 Hz), 7.45–7.27 (m, 5H), 7.21–7.00 (m, 5H), 5.83 (d, 1H, J=14.8 Hz), 4.16 (d, 1H, J=14.8 Hz), 3.38–3.15 (m, 2H), 3.00–2.81 (m, 2H), 1.82 (s, 3H); 13C-NMR (150 MHz, CDCl3) δ: 170.3, 140.5, 139.43, 139.37, 139.1, 135.4, 131.2, 130.0, 128.8, 128.7, 128.6, 128.54, 128.46, 127.8, 127.1, 126.9, 126.3, 52.8, 34.8, 31.2, 22.8; HR-MS (ESI) Calcd for C23H22NO (M+H+) 328.1696, Found: 328.1699.
5-Acetyl-8-bromo-5,6,11,12-tetrahydro-2-methylthiodibenz[b,f]azocine (25b)A screw top test tube equipped with a magnetic stirring bar was charged with mixture of 23b and 24b, Et3N (33 µL, 0.24 mmol) and anhydrous dichloromethane (0.50 mL). To the solution were added Ac2O (10 µL, 0.11 mmol) and DMAP (1.0 mg, 8.2 µmol) at room temperature. The solution was stirred for 1 h. The reaction was quenched with saturated aqueous NH4Cl, and the mixture was extracted with dichloromethane three times. The combined organic extracts were washed with brine, dried over anhydrous sodium sulfate, and filtered. The filtrate was concentrated under reduced pressure to leave the residue, which was purified by preparative TLC (hexanes–EtOAc=3 : 1) to afford acetamide 25b (12.4 mg, 0.033 mmol, 70%) and acetamide 27b (2.0 mg, 0.0053 mmol, 11%).
5-Acetyl-8-bromo-5,6,11,12-tetrahydro-2-methylthiodibenz[b,f]azocine (25b)A colorless oil; IR (neat) cm−1: 3002, 2921, 1657, 1491, 1386, 1286, 754; 1H-NMR (400 MHz, CDCl3) δ: 7.26–7.20 (m, 2H), 7.03 (dd, 1H, J=8.0, 2.0 Hz), 6.97 (d, 1H, J=8.0 Hz), 6.91 (d, 1H, J=8.0 Hz), 6.89 (d, 1H, J=2.0 Hz), 5.73 (d, 1H, J=14.8 Hz), 3.99 (d, 1H, J=14.8 Hz), 3.24–3.10 (m, 2H), 2.88–2.70 (m, 2H), 2.42 (s, 3H), 1.80 (s, 3H); 13C-NMR (100 MHz, CDCl3) δ: 170.3, 139.4, 139.3, 139.1, 137.3, 137.2, 132.7, 131.1, 130.8, 128.8, 128.4, 125.1, 119.8, 52.0, 34.7, 30.7, 22.7, 15.3; HR-MS (ESI) Calcd for C18H19BrNOS (M+H+) 376.0365, Found: 376.0381.
5-Acetyl-3-bromo-5,6,11,12-tetrahydro-9-methylthiodibenz[b,f]azocine (27b)A colorless oil; IR (neat) cm−1: 2292, 1660, 1587, 1487, 1382, 1295, 754; 1H-NMR (600 MHz, CDCl3) δ: 7.31–7.25 (m, 1H), 7.22 (d, 1H, J=1.8 Hz), 7.03 (d, 1H, J=7.2 Hz), 6.99–6.90 (m, 3H), 5.68 (d, 1H, J=15.0 Hz), 4.03 (d, 1H, J=15.0 Hz), 3.26–3.15 (m, 1H), 3.15–3.05 (m, 1H), 2.91–2.81 (m, 1H), 2.81–2.73 (m, 1H), 2.42 (s, 3H), 1.81 (s, 3H); 13C-NMR (100 MHz, CDCl3) δ: 169.8, 142.1, 140.6, 138.5, 138.0, 132.6, 131.7, 131.5, 131.4, 130.8, 127.6, 124.2, 120.3, 52.1, 34.2, 31.3, 22.8, 15.7; HR-MS (ESI) Calcd for C18H19BrNOS (M+H+), 376.0365; Found: 376.0351.
5-Acetyl-5,6,11,12-tetrahydro-2-methylthio-8-phenyldibenz[b,f]azocine (26)A screw top test tube equipped with a magnetic stirring bar was charged with aryl bromide 25b (35.2 mg, 0.0935 mmol), phenylboronic acid (23.2 mg, 0.190 mmol), K2CO3 (65.5 mg, 0.474 mmol), Pd(PPh3)4 (11 mg, 9.5 µmol), THF (0.4 mL), and H2O (0.1 mL). The mixture was heated to reflux for 22 h. The reaction mixture was diluted with EtOAc, washed with brine, dried over anhydrous sodium sulfate, and filtered. The filtrate was concentrated under reduced pressure to leave the residue, which was purified by preparative TLC (hexanes–EtOAc=3 : 1) to afford 26 (27.1 mg, 0.0726 mmol, 78%) as a colorless oil; IR (neat) cm−1: 3349, 2888, 1637, 1388, 1017, 762; 1H-NMR (400 MHz, CDCl3) δ: 7.51 (d, 2H, J=8.0 Hz), 7.43–7.26 (m, 5H), 7.11 (d, 1H, J=8.0 Hz), 7.04–6.96 (m, 2H), 6.91 (s, 1H), 5.82 (d, 1H, J=14.8 Hz), 4.13 (d, 1H, J=14.8 Hz), 3.35–3.12 (m, 2H), 2.94–2.80 (m, 2H), 2.40 (s, 3H), 1.81 (s, 3H); 13C-NMR (100 MHz, CDCl3) δ: 170.3, 140.5, 139.8, 139.2, 139.1, 138.9, 137.7, 135.4, 130.0, 128.8, 128.7, 128.6, 128.5, 127.1, 126.9, 126.4, 125.0, 52.7, 34.9, 31.0, 22.8, 15.3; HR-MS (ESI) Calcd for C24H24NOS (M+H+) 374.1573, Found: 374.1576.
5-Acetyl-5,6,11,12-tetrahydro-8-phenyldibenz[b,f]azocine (3)A Schlenk tube equipped with a magnetic stirring bar was charged with sulfide 26 (10.8 mg, 0.0289 mmol) and Raney nickel (W-2) in ethanol (0.5 mL). To the flask was charged with hydrogen gas (1 atm). The reaction mixture was heated to reflux for 3 h. The reaction mixture was filtered through a pad of Celite. The filter cake was washed with EtOAc, and the filtrate was concentrated under reduced pressure to leave the residue, which was purified by preparative TLC (hexanes–EtOAc=3 : 1) to afford 3 (6.6 mg, 0.020 mmol, 69%).