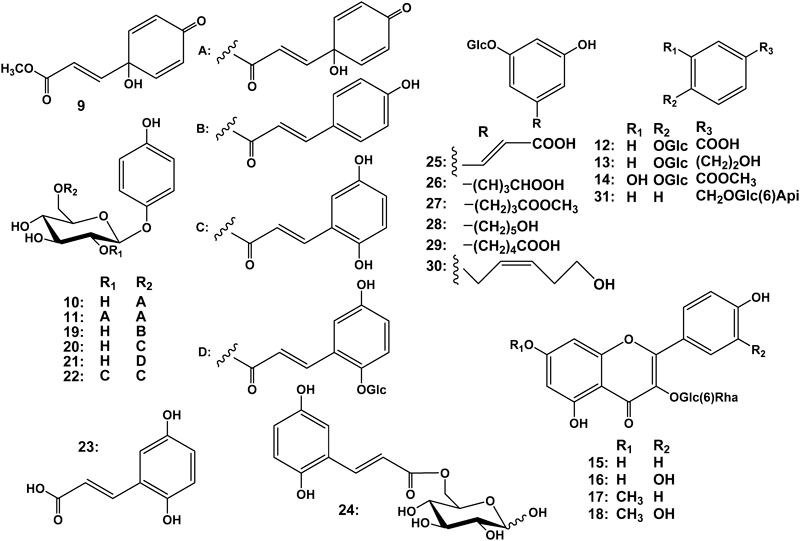
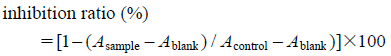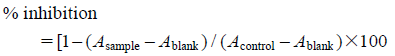Results and Discussion
Air-dried leaves of G. robusta were extracted with MeOH three times, followed by concentration. The MeOH extract was then partitioned with solvents of increasing polarity. The 1-BuOH-soluble fraction was separated by means of various chromatographic procedures comprising column chromatography (CC) procedures including a highly-porous synthetic resin CC (Diaion HP-20), normal silica gel CC, reversed-phase (octadecyl silica (ODS)) CC, droplet counter-current chromatography (DCCC), and HPLC to afford eight new compounds, named grevillosides J–Q (1–8), together with ten known compounds (9–18). The isolation details and yields are given in Experimental. The structures of the new compounds were elucidated mainly by spectroscopic evidence, and those of known phenolic compounds were identified as graviquinone (9),5) robustasides D (10)3) and G (11),6) p-hydroxybenzoic acid glucoside (12),7) icariside D2 (13),8) 4-O-β-D-glucopyranoside-3-hydroxy methyl benzoate (14),9) rutin (15),10) and rhamnocitrin (16),11) rhamnetin (17)12) and kaempferol 3-O-rutinosides (18)10) by comparison with data reported in the literature.
Grevilloside J (1), [α]D −45.8, was isolated as an amorphous powder and its elemental composition was determined to be C17H24O8 by high-resolution electrospray-ionization mass-spectrometry (HR-ESI-MS). The IR spectrum exhibited absorption bands ascribable to hydroxy groups (3381 cm−1) and a carbonyl functional group (1714 cm−1). In the 1H-NMR spectrum, two deshielded protons for an AA′BB′ spin–spin coupling system [δH 6.94 (2H, d, J=9 Hz) and 6.68 (2H, d, J=9 Hz) were observed. The heteronuclear single-quantum correlation (HSQC) spectrum revealed that these protons were at δC 119.8 and 116.6, respectively. Thus, these proton and carbon signals together with two other aromatic carbon signals (δC 152.3, 154.0) represented a hydroquinone skeleton that is present in robustaside D (10). An anomeric proton (δH 4.71) at δC 103.7 and five other oxygenated carbon signals (δC 78.0, 75.5, 75.0, 71.9, 64.9) indicated a sugar moiety. Acid hydrolysis of 1 gave D-glucose and the mode of sugar linkage was determined to be β from the coupling constant of the anomeric proton (J=8 Hz). A carbonyl carbon (δC 178.2) and the relatively deshielded methylene carbon (δC 64.9) of glucose indicated that 1 was an acylated arbutin derivative and the acyl moiety was expected to be a simpler compound with five carbon atoms. Other than the carbonyl carbon, the acyl moiety comprised two methyl, one methylene and one methine carbons. In the 1H-NMR spectrum, one methyl appeared as a triplet signal and another as a doublet signal. Based on the 1H–1H correlation spectroscopy (COSY) and the heteronuclear multiple-bond correlation (HMBC) spectra, the acyl moiety was assigned as 2-methylbutyrate and the esterification site was determined to be the hydroxy group at the 6′-position of the sugar moiety. The absolute configuration of the chiral center of methyl 2-methlybutyrate obtained on mild alkaline hydrolysis was confirmed to be S on HPLC analysis involving authentic methyl (+)-(2S)-2-methylbutyrate and a chiral detector. Therefore, the structure of 1 was elucidated to be that shown in Fig. 1.
Grevilloside K (2), [α]D −19.3, was isolated as an amorphous powder and its elemental composition was determined to be C30H28O13 by HR-ESI-MS. Grevilloside K (2) was expected to be an arbutin derivative with different acyl groups and thus the IR spectrum exhibited absorption bands ascribable to two types of carbonyl functional groups (1669, 1704 cm−1). From the NMR spectroscopic data, the acyl groups were assigned to be 2,5-dihydroxycinnamic acid, which is present in robustasides B and C4) (B in Fig. 1) and (E)-3-(1-hydroxy-4-oxocyclohexa-2,5-dien-1-yl)acrylate which is present in robustasides D3) (C in Fig. 1). Acid hydrolysis of 2 gave D-glucose and the mode of sugar linkage was determined to be β from the coupling constant of the anomeric proton (J=8 Hz). The positions of acyl substituents were confirmed to be the hydroxy groups at the 2′- and 6′-positions on two-dimensional NMR spectroscopy. In the COSY spectrum, the anomeric proton signal (H-1′) was correlated with a deshielded signal at δH 5.05 (H-2′), which showed a correlation cross peak with δC 168.6 in the HMBC spectrum. Further HMBC correlations shown in Fig. 3 established the carbonyl carbon at δC 168.6 was that of the 2,5-dihydroxycinnamate, and another carbonyl carbon at δC 167.2 showed HMBC correlation peaks with H2-6′ (δH 4.37, 4.54). Therefore, the structure of 2 was elucidated to be robustaside D 2′-O-2,5-dihydroxycinnamate, as shown in Fig. 1.

Fig. 3. 1H–1H COSY and HMBC Correlations for 2
Grevillosides L (3), [α]D −44.6, was isolated as an amorphous powder and its elemental composition was determined to be C21H24O11 by HR-ESI-MS. The IR spectrum exhibited absorption assignable to hydroxyl groups (3391 cm−1), two types of carbonyl functional groups (1712, 1674 cm−1), and an aromatic ring (1509 cm–1). Of the 19 significant signals observed in the 13C-NMR spectrum, some appeared as two extremely close resonances. An arbutin moiety (C-1 to C-6 and C-1′ to C-6′) was obviously present in the molecule and in the 13C-NMR spectrum, the C6–C3 acyl moiety comprised trans and cis double bonds, two carbonyl, methylene, oxygenated methylene and methine carbons. In the 1H–1H COSY spectrum, methylene protons and an oxymethine proton showed a cross peak, and the oxymethine proton further correlated with one of the olefinic protons at the cis double bond due to a W-figure formation. Based on the latter together with the evidence obtained from the HMBC spectrum (Fig. 4), the structure of the acyl moiety was elucidated to be a H2O adduct of (E)-3-(1-hydroxy-4-oxocyclohexa-2,5-dien-1-yl)acrylate (C in Fig. 1), namely (E)-3-(1,6-dihydroxy-4-oxocyclohex-2-en-1-yl)acrylate (D in Fig. 1). There are two hydroxylation sites, i.e., the 2″- and 6″-positions of C, and the regioselectivity between these positions seemed to be low. Judging from the formation of the W-figure between H-2″ and H-6″, and the significant nuclear Overhauser effect (NOE) correlation between H2-5″ and H-7″ (Fig. 5), two hydroxy groups at the 1″- and 6″-positions of D must be directed in the same orientations. Therefore, the hydroxylation must occur at either the si-face of the pro-R (2″)-position or the re-face of the pro-S (6″)-position, enantiomeric 1-R, 6-R and 1-S, 6-S isomers being formed, respectively. As a result, grevillosides L (3) was isolated as an inseparable diastereomeric mixture. Although several different columns [Cosmosil-Cholester and -Hilic (Nakalai Tesque), Develosil C30 (Nomura Chemical), etc.] and solvent systems were tried to separate them, probably due to the remoteness of the chiral centers in the acyl group and the 5′-position, this mixture could not be separated into single compounds.

Fig. 4. 1H–1H COSY and HMBC Correlations for 3
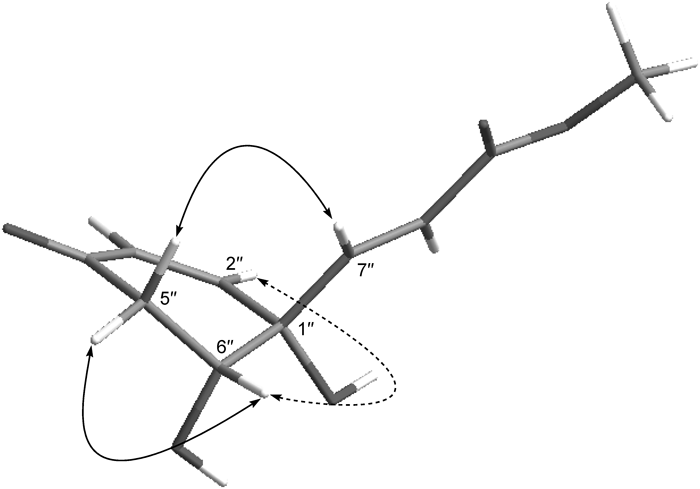
Fig. 5. The Solid Lines Indicate NOE Correlations and the Dotted Line a Long-Range Correlation of 3
Grevillosides M (4), [α]D −38.5, was also isolated as an inseparable mixture like 3 and its elemental composition was determined to be C22H26O11 by HR-ESI-MS. The NMR spectroscopic data showed closely resembled those of 3, except for the appearance of methoxy signals at δH 3.41 and 3.42 at δC 58.44, which corresponds the increase of 14 mass unit, when compared with 3 (Table 1). Other spectroscopic data were essentially the same as those of 3. The stereochemistry of C-1″ and 6″ was the same as that in 3, from a W-figure long-range coupling between H-2″ and H-6″, observed in the 1H-NMR spectrum and also a NOE correlation, observed between H2-5″ and H-7″. Therefore, the structure of 4 was elucidated to be as shown in Fig. 1. This methoxy derivatives may be formed during extraction and separation procedures.
Table 1.
13C-NMR Spectroscopic Data for Grevillosides J–Q (
1–
8) and Robustaside D (
10) (100 MHz, CD
3OD)
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 10 |
|---|
| 1 | 152.3 | 152.1 | 152.29 | 152.28 | 152.24 | 152.2 | 152.6 | 152.3 | | | 152.3 |
| 2 | 119.8 | 119.8 | 119.66 | 119.62 | 119.63 | 119.6 | 106.9 | 119.3 | | | 119.7 |
| 3 | 116.6 | 116.8 | 116.75 | 116.74 | 116.7 | 146.8 | 116.8 | | | 116.8 |
| 4 | 154.0 | 154.3 | 153.96 | 153.99 | 154.0 | 142.0 | 153.8 | | | 154.0 |
| 5 | 116.6 | 116.8 | 116.75 | 116.74 | 116.7 | 116.4 | 116.8 | | | 116.8 |
| 6 | 119.8 | 119.8 | 119.66 | 119.62 | 119.63 | 119.6 | 109.2 | 119.3 | α | β | 119.7 |
| 1′ | 103.7 | 102.2 | 103.70 | 103.67 | 103.68 | 103.64 | 103.7 | 103.5 | 102.2 | 94.1 | 98.3 | 103.7 |
| 2′ | 75.0 | 75.2 | 74.98 | 75.0 | 75.0 | 74.9 | 83.9 | 73.9 | 76.3 | 75.0 |
| 3′ | 78.0 | 76.1 | 77.95 | 78.0 | 77.9 | 78.0 | 77.1 | 74.9 | 78.0 | 78.0 |
| 4′ | 71.9 | 71.9 | 71.83 | 71.9 | 71.7 | 71.8 | 71.2 | 72.1 | 71.8 | 71.8 |
| 5′ | 75.5 | 75.5 | 75.37 | 75.34 | 75.35 | 75.31 | 75.3 | 75.3 | 77.75 | 70.7 | 75.4 | 75.3 |
| 6′ | 64.9 | 65.0 | 65.07 | 65.04 | 65.1 | 65.0 | 65.1 | 67.7 | 65.3 | 65.4 | 65.1 |
| 1″ | 178.2 | 123.0 | 73.78 | 73.76 | 73.94 | 80.1 | 70.4 | 105.5 | 70.4 | 70.4 |
| 2″ | 42.5 | 151.6 | 150.19 | 150.17 | 150.55 | 150.49 | 146.8 | 151.2 | 76.1 | 151.1 | 151.2 |
| 3″ | 27.9 | 118.1 | 130.55 | 130.53 | 130.43 | 130.38 | 130.7 | 128.7 | 77.81 | 128.7 | 128.7 |
| 4″ | 12.0 | 120.4 | 199.44 | 199.42 | 198.73 | 196.3 | 187.1 | 71.3 | 187.0 | 187.7 |
| 5″ | 16.9 | 151.4 | 43.40 | 40.06 | 32.3 | 128.7 | 77.9 | 128.7 | 128.7 |
| 6″ | | 114.9 | 73.78 | 73.76 | 83.04 | 82.99 | 49.9 | 151.2 | 62.6 | 151.1 | 151.2 |
| 7″ | | 142.7 | 150.15 | 149.50 | 149.41 | 148.2 | 148.2 | | 148.14 or 148.19 | 148.2 |
| 8″ | | 118.0 | 123.32 | 123.30 | 123.38 | 123.35 | 122.5 | 122.9 | | 122.87 or 122.93 | 122.9 |
| 9″ | | 168.6 | 167.46 | 167.37 | 167.1 | 167.3 | | 167.3 | 167.2 |
| –OCH3 | | | | | 58.44 | | | | | | |
| 1‴ | | 70.4 | | | | | 177.4 | | 102.3 | | | |
| 2‴ | | 151.2 | | | | | 79.1 | | 72.1 | | | |
| 3‴ | | 128.7 | | | | | 106.9 | | 72.4 | | | |
| 4‴ | | 187.0 | | | | | 84.8 | | 74.2 | | | |
| 5‴ | | 128.7 | | | | | 70.9 | | 69.8 | | | |
| 6‴ | | 151.2 | | | | | 62.8 | | 18.1 | | | |
| 7‴ | | 148.2 | | | | | | | | | | |
| 8‴ | | 122.9 | | | | | | | | | | |
| 9‴ | | 167.2 | | | | | | | | | | |
Grevilloside N (5), [α]D −76.0, was isolated as an amorphous powder and its elemental composition was determined to be C27H30O16. The IR spectrum exhibited absorption assignable to hydroxy groups (3346 cm−1), γ-lactone (1775 cm−1), and two types of conjugated ketones (1715, 1677 cm−1). NMR spectroscopic data indicated that grevilloside N (5) was an analogous compound to previous ones, namely, an arbutin moiety is present in the molecule and the hydroxy group at the 6′-position was also acylated. However, the acyl moiety comprised not nine but 15 carbons. Signals for a trans double bond [δH 6.17 (d, J=16 Hz) and 7.06 (d, J=16 Hz)] and an AB doublet [δH 5.95 (d, J=10 Hz) and 6.49 (dd, J=10, 2 Hz)] were observed in the 1H-NMR spectrum, and the 13C-NMR spectrum exhibited an oxygenated tertiary carbon at a fairly deshielded position (δC 80.1) and a highly deshielded carbonyl carbon signal (δC 196.3). Although these signals were assignable to the partial structure of (E)-3-(1-hydroxy-4-oxocyclohexa-2,5-dien-1-yl)acrylate, the AA′BB′ doublet was replaced by an AB doublet. The 13C-NMR spectrum further revealed that the remaining signals comprised those for methylene, methine, ketal, carbonyl, oxygenated tertiary, two oxygenated secondary and oxygenated primary carbons. These signals indicated that one of the double bonds in the ring of (E)-3-(1-hydroxy-4-oxocyclohexa-2,5-dien-1-yl)acrylic acid was involved in the modification with six carbon atoms. In the 1H–1H COSY spectrum, two prominent proton sequences, i.e., H2-5″ to H-6″ and H-4‴ to H-6‴ via H-5‴, were observed along with long-range coupling between H-2″ and H-6″ (Fig. 6), which indicated that one of the double bonds in the ring was converted to be one methylene and one methine during the building of a new skeleton. The HMBC correlations from H-4‴ (δH 4.67) to C-1‴ (δC 177.4), C-2‴ (δC 79.1), and C-3‴ (δC 106.9) established the involvement of C-1‴ to C-4‴ in the γ-lactone ring formation, and those from H-5″ (δH 2.68, 3.09) to C-2‴ and H-6″ (δH 3.18) to C-1‴ substantiated the connectivity of the γ-lactone ring and the six-membered ring at the C-6″ and C-2‴ positions. The elemental composition demands one more degree of unsaturation and this was satisfied by the formation of a cyclic system sharing an oxygen atom between C-1″ and C-3‴. The significant downfield shift of C-1″ (δC 80.1) from δC 70.4 in 2 in the 13C-NMR spectra and the presence of a stable hemiketal also supported this finding. From the biosynthetic point of view, grevilloside N (5) was probably formed through the cycloaddition of ascorbic acid toward the (E)-3-(1-hydroxy-4-oxocyclohexa-2,5-dien-1-yl)acrylate of robustaside D (10) (Fig. 2). As to the stereochemistry, the C-4‴ and C-5‴ positions may be the same as those of ascorbic acid and the hydroxy groups at the ring junctions of five-membered rings may be in anti-parallel positions, but the absolute stereochemistry is remained uncertain. The bonds, 1″ to the ketal oxygen and 6″ to 2‴, must be orientated in the same direction, otherwise, H-2″ and H-6″ could not adopt a W-figure plain to induce a long-range coupling. Therefore, the structure of grevilloside N was tentatively elucidated to be 5 shown in Fig. 1.

Grevilloside O (6), [α]D −15.4, was isolated as an amorphous powder and its elemental composition was determined to be C21H22O11. In the NMR spectra, typical signals assignable to (E)-3-(1-hydroxy-4-oxocyclohexa-2,5-dien-1-yl)acrylate and those of 6-acylated β-D-glucopyranoside were observed, as seen in previous compounds. Although the aglycone moiety comprised six carbons, it was asymmetrically substituted by three oxygen atoms. The three aromatic proton signals were coupled in an ABX system, and in the difference NOE experiment, on irradiation of the anomeric proton at δH 4.70 (d, J=7 Hz), significant signal enhancements were observed for H-2 [δH 6.58 (d, J=3 Hz) and H-6 [δH 6.42 (dd, J=9, 3 Hz)]. Therefore, the structure of 6 was elucidated to be 1,3,4-trihydroxybenzene 1-O-β-D-glucopyranoside 6′-O-(E)-3-(1-hydroxy-4-oxocyclohexa-2,5-dien-1-yl)acrylate, as shown in Fig. 1.
Grevilloside P (7), [α]D −47.1 was isolated as an amorphous powder and its elemental composition was determined to be C24H36O16 by HR-ESI-MS. The IR spectrum exhibited absorption bands ascribable to hydroxy groups (3357 cm−1) and an aromatic ring (1510 cm−1). NMR spectral data suggested that 7 is also a congeneric compound to arbutin. Three anomeric protons signals (δH 4.62, 4.67, 4.91) observed in the 1H-NMR spectrum showed correlation cross peaks with the respective anomeric carbon signals (δC 105.5, 102.3, 102.2) in the HSQC spectrum. Since the 13C-NMR spectrum exhibited twelve signals ascribable to terminal α-rhamnopyranoside and β-glucopyranoside, and the sugar analysis revealed the presence of L-rhamnose and D-glucose, the inner hexose was expected to be β-D-glucopyranose attached to p-hydroquinone aglycone. Acetylation of 7 gave decaacetate (7a), and thus the enormously overlapping 1H-NMR signals observed in the sugar region became clear as well-resolved peaks. One of the anomeric protons at δH 5.19 (d, J=8 Hz) showed correlation with the highly deshielded aromatic carbon at δC 156.1 (C-1) in the HMBC spectrum and it also showed a correlation peak signal with intact δH 3.90 at δC 78.7 (C-2′) in the 1H–1H COSY spectrum. The anomeric proton signal at δH 4.93 (1H, d, J=8 Hz) was expected to be that of the terminal glucopyranoside and its HMBC correlation to C-2″ established one of the sugar linkages to be Glc(1″→2′)Glc. The HMBC correlation between the anomeric proton of the terminal rhamnopyranoside at δH 5.35 (1H, d, J=2 Hz, H-1‴) and δC 66.5 with intact methylene protons (δH 3.63, 3.72) revealed the remaining sugar linkage to between rhamnopyranose and the 6-position of either glucopyranose. Since the 1H–1H COSY spectrum clearly demonstrated the sequences of the sugar ring protons and those at the 6-positions of both glucopyranoses, attachment of the terminal rhamnopyranose was determined to be at the 6′-position of the inner glucopyranose, as shown in Fig. 1.
Grevilloside Q (8), [α]D +16.3, was isolated as an amorphous powder and its elemental composition was determined to be C15H18O9 by HR-ESI-MS. On HPLC separation, 8 appeared as two interconvertible peaks and the presence of D-glucose was confirmed by HPLC analysis of the hydrolyzate. In the NMR spectra, two anomeric resonances were observed at δH 4.48 on δC 98.3 and δH 5.08 on δC 94.1, the former being assigned as that of the β-anomer and the latter as that of the α-anomer from their coupling constants, 8 Hz and 4 Hz, respectively. Some of the NMR signals for the acyl group appeared as two close signals and the acyl moiety was assigned as (E)-3-(1-hydroxy-4-oxocyclohexa-2,5-dien-1-yl)acrylate (C in Fig. 1). The abundance ratio of α- and β-anomers was estimated to be nearly 1 : 1 from the integrals of isolated signals in the 1H-NMR spectrum.
Sun burn and chloasma are caused by the deposition of melanin pigment, which is formed on sun light stimulation of melanocytes located in the basement membrane. Arbutin is a natural skin-whitening agent originally isolated from bearberry. Arbutin itself and synthetic α-arbutin (hydroquinone α-D-glucopyranoside) are available on the market as skin-whitening agents and they inhibit tyrosinase, which is involved in the early stage of melanin production.13) G. robusta was found to be a rich source of arbutin derivatives in our study. The compounds (1–18) isolated in this study together with those (19–31) in our previous study1,2,4) were assayed as to their tyrosinase and melanogenesis inhibitory activity by using mouse melanoma cells (B16). However, none of the compounds tested showed any tyrosinase inhibitory activity (positive control, kojic acid: IC50=17.9±2.4 µM). Chuang et al. reported a similar result for graviquinone (9), i.e., it exhibited almost no tyrosinase inhibitory activity (32.78±0.54% inhibition at the concentration of 16.7 g/mL).5) On the other hand, of the 31 compounds, 5-alkylresorcinol derivatives (25–30), phenolic compounds (12, 14, 23, 24, 31), and flavonoid glycosides (15–18) were also inactive as to B16 melanogenesis, exception being 13 (Icariside D2) (59.8% inhibition at 100 µM). The melanogenesis inhibitory activity of icariside D2 has already been reported in a patent document.14) Significant activity was exclusively observed for arbutin derivatives (1–11, 19–22) using B16 melanoma cells (2, 59.9±3.0%: 4, 74.3±4.6%: 10, 45.9±6.5%: 19, 55.2±4.0%, and arbutin, 34.3±2.5% inhibition at 100 µM (3, 91.9% at 25 µM, and 9, 56.6% at 1.6 µM). These preliminary data were further confirmed using a high-melanin producing clone, B16Y24, established in this study (Table 2). Although B16Y24 is a potent melanin producer, grevillosides K and O (2, 6), and robustasides D (10) and G (11) could inhibit melanogenesis moderately, and grevillosides M (4) and graviquinone (9) possessed potent inhibitory activity toward it (Table 2). It is noteworthy that their strong melanogenesis inhibitory activity showed almost no association with cytotoxicity (Table 2). Considering the structures and activity relationship, these compounds possessed a common ester moiety i.e., 3-(1-hydroxy-4-oxocyclohexa-2,5-dien-1-yl)acrylate or (E)-3-(1,6-dihydroxy-4-oxocyclohex-2-en-1-yl)acrylate. Therefore, graviquinone (9), the ester moiety of grevillosides M (4), and presumably its methyl ester are relatively small molecules that can be produced with synthetic easiness, and thus they are promising compounds for skin-lightening and anti-chloasma agents.
Table 2. Melanogenesis Inhibitory Activity (B16Y24)
| Melanogenensis IC50 (µM) | Cytotoxicity IC50 (µM) |
|---|
| 2 | >30a) | >30 |
| 4 | 7.5±3.1 | >30 |
| 6 | 52.9±2.5b) | >100b) |
| 9 | 11.3±0.1 | >30 |
| 10 | 20.7±1.8 | >30 |
| 11 | >30c) | >30 |
| Arbutin | 175.1±3.4b) | >300b) |
Each value represents the mean±S.D. for quadruple experiments. a, c) 45.2±7.3 and 34.7±3.0%, respectively, with the highest dose of 30 µM, due to the limited quantities of samples available. b) The dose was increased up to 100 or 300 µM to determine the IC50 values.
Experimental
General Experimental ProcedureOptical rotations were measured on a JASCO P-1030 digital polarimeter. IR and UV spectra were measured on Horiba FT-710 and JASCO V-520 UV/Vis spectrophotometers, respectively. 1H- and 13C-NMR spectra were taken on a JEOL JNM α-400 at 400 MHz and 100 MHz, respectively, with tetramethylsilane as an internal standard. HR-ESI-MS was performed with an Applied Biosystems QSTAR XL NanoSpray™ System.
A highly-porous synthetic resin (Diaion HP-20) was purchased from Mitsubishi Kagaku (Tokyo, Japan). Silica gel CC was performed on silica gel 60 (E. Merck, Darmstadt, Germany), and ODS open CC on Cosmosil 75C18-OPN (Nacalai Tesque, Kyoto, Japan) [Φ=50 mm, L=25 cm, linear gradient: MeOH–H2O (1 : 9, 1 L)→(1 : 1, 1 L), fractions of 10 g being collected]. The DCCC (Tokyo Rikakikai, Tokyo, Japan) was equipped with 500 glass columns (Φ=2 mm, L=40 cm), the lower and upper layers of a solvent mixture of CHCl3–MeOH–H2O–1-PrOH (9 : 12 : 8 : 2) being used as the stationary and mobile phases, respectively. Five-gram fractions were collected and numbered according to their order of elution with the mobile phase. HPLC was performed on an ODS column (Inertsil; GL Science, Tokyo, Japan; d=6 mm, L=250 mm, 1.6 mL/min), and the eluate was monitored with a UV detector at 254 nm and a refractive index monitor. (S)-(+)-2-Methylbutanoic acid was purchased from Wako Pure Chemical Industries, Ltd. (Osaka, Japan) and mushroom tyrosinase was obtained from Sigma-Aldrich Corporation (St. Louis, MO, U.S.A.).
Plant MaterialThe leaves of G. robusta were collected in Nakagami-gun, Okinawa in June 2005, A voucher specimen was deposited in the Herbarium of the Department of Pharmacognosy, Graduate School of Biomedical and Health Sciences, Hiroshima University (05-GR-Okinawa-0629).
Extraction and IsolationDried leaves of G. robusta (6.37 kg) were extracted three times with MeOH (30 L) at 25°C for one week, followed by concentration to 3 L in vacuo. The extract was washed with n-hexane (3 L, 32.6 g) and then the MeOH layer was concentrated to a gummy mass. The latter was suspended in water (3 L) and then extracted with EtOAc (3 L) to give 160 g of an EtOAc-soluble fraction. The aqueous layer was extracted with 1-BuOH (3 L) to give a 1-BuOH-soluble fraction (405 g), and the remaining water layer was concentrated to yield 475 g of a water-soluble fraction. A portion (237 g) of the 1-BuOH-soluble fraction was applied to a Diaion HP-20 column (Φ=75 mm, L=50 cm) using H2O–MeOH (4 : 1, 6 L), (2 : 3, 6 L), (3 : 2, 6 L), (1 : 4, 6 L), and MeOH (6 L), 1 L fractions being collected. The residue (19.9 g) in fractions 4–6 of the 20% MeOH eluent was subjected to silica gel (450 g) CC, with elution with CHCl3 (3 L) and CHCl3–MeOH [(49 : 1, 3 L), (24 : 1, 3 L), (23 : 2, 3 L), (9 : 1, 3 L), (7 : 1, 3 L), (17 : 3, 3 L), (4 : 1, 3 L), (3 : 1, 3 L), and (3 : 2, 3 L)], 500 mL fractions being collected. The residue (2.50 g) in fractions 42–45 of the 15–20% MeOH eluate was separated by ODS open CC to give a residue in fractions 34–38 (461 mg), followed by separation by DCCC to yield a residue (198 mg) in fractions 20–27, which was purified by HPLC (H2O–MeOH, 20 : 3) to give 8 (6.8 mg) and 13 (4.0 mg). Combined fractions 46–52 (3.84 g) of the 20–25% MeOH eluate were separated by ODS open CC and then DCCC to afford 12 (9.7 mg) and 14 (12.8 mg). An aliquot (2.19 g) of combined fractions 53–66 (3.87 g) of the 25–40% MeOH eluate was separated by ODS open CC and the residue (81.8 mg) in fractions 35–44 by DCCC to give 31.0 mg of a residue in fractions 10–15. The latter was purified by HPLC (H2O–MeOH, 9 : 1) to give 11.8 mg of 7 from the peak at 11.5 min.
The precipitate in fractions 7–12 of the 20–40% MeOH eluent on Diaion HP-20 CC was collected by filtration to afford 642 mg of 16. The remaining residue (44.5 g) was subjected to silica gel (500 g) CC, with elution with CHCl3 (4.5 L) and CHCl3–MeOH [(49 : 1, 4.5 L), (24 : 1, 4.5 L), (19 : 1, 4.5 L), (23 : 2, 4.5 L), (9 : 1, 4.5 L), (7 : 1, 4.5 L), (17 : 3, 4.5 L), (33 : 7, 4.5 L), (4 : 1, 4.5 L), (3 : 1, 4.5 L), (7 : 3, 4.5 L)], 500 mL fractions being collected. An aliquot (1.87 g) of combined fractions 48–52 (13.1 g) of the 8–10% MeOH eluate was separated by ODS open CC to give two residues in fractions 83–96 (682 mg) and 312 mg of 10 in fractions 109–124. The former was separated by DCCC to give 620 mg of 10 in fractions 41–51 and 75.4 mg of 4 in fractions 52–58. An aliquot (1.80 g) of combined fractions 53–60 (8.79 g) of the 10–12.5% MeOH eluate was separated by ODS open CC to give two residues in fractions 66–80 (164 mg) and fractions 89–99 (544 mg). The former was separated by DCCC to give 43.8 mg of 4 in fractions 17–25 and the latter to afford 433 mg of 10 in fractions 35–43. An aliquot (1.76 g) of combined fractions 61–66 (2.61 g) of the 12.5–15% MeOH eluate was separated by ODS open CC to give two residues in fractions 69–87 (176 mg) and 88–99 (204 mg). The former was separated by DCCC to give 70.0 mg of 3 in fractions 17–22, and the latter to give a residue in fractions 25–28 (38.5 mg), which was purified by HPLC by (H2O–MeOH, 3 : 1) to give 8.8 mg of 6 from the peak at 13 min. An aliquot (1.85 g) of combined fractions 67–73 (2.01 g) of the 15–17.5% MeOH eluate was separated by ODS open CC and the residue (458 mg) in fractions 100–115 by DCCC to give 130 mg in fractions 15–22 and the latter was then purified by HPLC (H2O–MeOH, 4 : 1) to give 18.8 mg of 5 from the peak at 45 min.
The residue (39.0 g in fractions 13–18) of the 40–60% MeOH eluate obtainedon Diaion HP-20 CC was subjected to silica gel (450 g) CC, with elution with CHCl3 (4.5 L) and CHCl3–MeOH [(49 : 1, 4.5 L), (24 : 1, 4.5 L), (19 : 1, 4.5 L), (23 : 2, 4.5 L), (9 : 1, 4.5 L), (7 : 1, 4.5 L), (17 : 3, 4.5 L), (33 : 7, 4.5 L), (4 : 1, 4.5 L), (3 : 1, 4.5 L), (7 : 3, 4.5 L)], 500 mL fractions being collected. An aliquot (1.80 g) of combined fractions 40–47 (10.2 g) of the 8–10% MeOH eluate was separated by ODS open CC to give a residue in fractions 147–164 (1.04 g). The latter was separated by DCCC to give a residue in fractions 83–93 (20.3 mg) and 224 mg of 11 in fractions 99–108. The former was purified by HPLC (H2O–MeOH, 1 : 1) to give 9.3 mg of 1 from the peak at 9 min. The residue (1.90 g) in fractions 48–54 of the 10% MeOH eluate was separated by ODS open CC to give two residues in fractions 158–170 (525 mg) and fractions 171–180 (297 mg). The former was separated by DCCC to give a residue in fraction of 92–114 (264 mg), which was then purified by HPLC (H2O–MeOH, 7 : 3) to give 2.0 mg of 9 from the peak at 12 min. The latter was separated by DCCC to give a residue in fractions 41–51 (153 mg), which was then purified by HPLC (H2O–MeOH, 11 : 3) to give 11.8 mg of 2 from the peak at 8 min. An aliquot (1.82 g) of combined fractions 63–72 (3.58 g) of the 15% MeOH eluate was separated by ODS open CC to give 15 in fractions 175–189 (399 mg). The latter was separated by DCCC to give a residue in fractions 83–93 (20.3 mg) and 224 mg of 11 in fractions 99–108.
The residue (23.9 g in fractions 19–24) of the 60–80% MeOH eluate obtained on Diaion HP-20 CC was subjected to silica gel (400 g) CC, with elution with CHCl3 (3 L) and CHCl3–MeOH [(98 : 2, 3 L), (24 : 1, 3 L), (19 : 1, 3 L), (23 : 2, 3 L), (9 : 1, 3 L), (7 : 1, 3 L), (17 : 3, 3 L), (33 : 7, 3 L), (4 : 1, 3 L), (3 : 1, 3 L), (7 : 3, 3 L)], 500 mL fractions being collected. An aliquot (1.83 g) of combined fractions 40–49 (4.08 g) of the 15–20% MeOH eluate was separated by ODS open CC to give 17 in fractions 227–243 (111 mg). An aliquot (2.75 g) of combined fractions 50–57 (2.93 g) of the 20–30% MeOH eluate was separated by ODS open CC to give a residue in fractions 224–241 (601 mg). The latter was separated by DCCC to give 81.4 mg of 18 in fractions 41–51.
Grevilloside J (1)Amorphous powder; [α]D27 −45.8 (c=0.62, MeOH); IR νmax (film) cm−1: 3381, 2933, 1714, 1649, 1510, 1456, 1361, 1213, 1072, 1016; UV λmax (MeOH) nm (log ε): 285 (3.43), 222 (3.89); 1H-NMR (400 MHz, CD3OD) δ: 6.94 (2H, d, J=9 Hz, H-2, 6), 6.68 (2H, d, J=9 Hz, H-3, 5), 4.71 (1H, d, J=8 Hz, H-1′), 4.46 (1H, dd, J=12, 2 Hz, H-6′a), 4.18 (1H, dd, J=12, 6 Hz, H-6′b), 3.57 (1H, ddd, J=9, 6, 2 Hz, H-5′), 3.44 (1H, dd, J=9, 9 Hz, H-3′), 3.41 (1H, dd, J=9, 8 Hz, H-2′), 3.33 (1H, dd, J=9, 9 Hz, H-4′), 2.40 (1H, sextet, J=7 Hz, H-2″), 1.66 (1H, dqui, J=14, 7 Hz, H-3″a), 1.47 (1H, dqui, J=14, 7 Hz, H-3″b), 1.13 (3H, d, J=7 Hz, H3-5″), 0.89 (3H, t, J=7 Hz, H3-4″); 13C-NMR (100 MHz, CD3OD): Table 1; HR-ESI-MS (positive-ion mode) m/z: 379.1364 [M+Na]+ (Calcd for C17H24O8Na: 379.1363).
Grevilloside K (2)Amorphous powder. [α]D24 −19.3 (c=0.79, MeOH). IR νmax (film) cm−1: 3354, 2955, 1704, 1669, 1207, 1175, 1077. UV λmax (MeOH) nm (log ε): 279 (4.27), 222 (4.49). 1H-NMR (400 MHz, CD3OD) δ: 8.00 (1H, d, J=16 Hz, H-7″), 6.92 (1H, br s, H-6″), 6.85 (2H, d, J=10 Hz, H-2‴, 6‴), 6.83 (2H, d, J=9 Hz, H-2, 6), 6.71 (1H, d, J=16 Hz, H-7‴), 6.71 (2H, m, H-2″, 3″), 6.66 (1H, d, J=9 Hz, H-3, 5), 6.56 (1H, d, J=16 Hz, H-8″), 6.30 (1H, d, J=16 Hz, H-8‴), 6.23 (2H, d, J=10 Hz, H-3‴, 5‴), 5.05 (1H, dd, J=9, 8 Hz, H-2′), 4.95 (1H, d, J=8 Hz, H-1′), 4.54 (1H, dd, J=12, 2 Hz, H-6′a), 4.37 (1H, dd, J=12, 6 Hz, H-6′b), 3.71 (1H, ddd, J=9, 6, 2 Hz, H-5′), 3.70 (1H, dd, J=9, 9 Hz, H-3′), 3.49 (1H, dd, J=9, 9 Hz, H-4′). 13C-NMR (100 MHz, CD3OD): Table 1. HR-ESI-MS (positive-ion mode) m/z: 619.1396 [M+Na]+ (Calcd for C30H28O13Na: 619.1422).
Grevillosides L (3)Amorphous powder; [α]D22 −44.6 (c=0.28, MeOH); IR νmax (film) cm−1: 3391, 2920, 2868, 1712, 1674, 1509, 1214, 1062, 1016; UV λmax (MeOH) nm (log ε): 280 (3.45), 219 (4.18); 1H-NMR (400 MHz, CD3OD) δ: 7.03 (2H, d, J=16 Hz, H2-7″), 6.915 (2H, d, J=9 Hz, H-2a, 6a); 6.910 (2H, d, J=9 Hz, H-2b, 6b), 6.693 (2H, d, J=9 Hz, H-3a, 5a), 6.688 (2H, d, J=9 Hz, H-3b, 5b), 6.63 (2H, dd, J=10, 1 Hz, H2-2″), 6.24 (2H, d, J=16 Hz, H2-8″), 6.07 (2H, d, J=10 Hz, H2-3″), 4.72 (2H, d, J=7 Hz, H2-1′), 4.51 (2H, dd, J=12, 2 Hz, H2-6′a), 4.334 (1H, dd, J=12, 6 Hz, H-6′ba), 4.327 (1H, dd, J=12, 6 Hz, H-6′bb), 4.04 (2H, td, J=5, 1 Hz, H2-6″), 3.624 (1H, ddd, J=9, 6, 2 Hz, H-5′a), 3.619 (1H, ddd, J=9, 6, 2 Hz, H-5′b), 3.45 (2H, dd, J=9, 9 Hz, H-3′), 3.41 (2H, dd, J=9, 7 Hz, H-2′), 3.37 (2H, dd, J=9, 9 Hz, H-4′), 2.69 (4H, d, J=5 Hz, H4-5″); 13C-NMR (100 MHz, CD3OD): Table 1; HR-ESI-MS (positive-ion mode) m/z: 475.1204 [M+Na]+ (Calcd for C21H24O11Na: 475.1210).
Grevillosides M (4)[α]D22 −38.5 (c=0.47, MeOH); IR νmax (film) cm−1:3398, 2912, 2838, 1712, 1673, 1509, 1216, 1076, 1017; UV λmax (MeOH) nm (log ε): 282 (3.45), 219 (4.39); 1H-NMR (400 MHz, CD3OD) δ: 7.03 (2H, d, J=16 Hz, H2-7″), 6.913 (2H, d, J=9 Hz, H-2a, 6a); 6.908 (2H, d, J=9 Hz, H-2b, 6b), 6.690 (2H, d, J=9 Hz, H-3a, 5a), 6.685 (2H, d, J=9 Hz, H-3b, 5b), 6.59 (2H, dd, J=10, 1 Hz, H2-2″), 6.25 (2H, d, J=16 Hz, H2-8″), 6.04 (2H, d, J=10 Hz, H2-3″), 4.72 (2H, d, J=7 Hz, H2-1′), 4.51 (2H, dd, J=12, 2 Hz, H2-6′a), 4.334 (12H, dd, J=12, 6 Hz, H2-6′ba), 4.327 (1H, dd, J=12, 6 Hz, H-6′bb), 3.68 (2H, td, J=3, 1 Hz, H2-6″), 3.624 (1H, dd, J=9, 6, 2 Hz, H-5′a), 3.619 (1H, ddd, J=9, 6, 2 Hz, H-5′b), 3.42 (2H, dd, J=9, 9 Hz, H-3′), 3.42 (3H, s, CH3O-), 3.41 (2H, dd, J=9, 7 Hz, H-2′), 3.41 (3H, s, CH3O-), 3.37 (2H, dd, J=9, 9 Hz, H-4′), 2.75 (4H, d, J=3 Hz, H4-5″); 13C-NMR (100 MHz, CD3OD): Table 1; HR-ESI-MS (positive-ion mode) m/z: 489.1369 [M+Na]+ (Calcd for C22H26O11Na: 489.1367).
Grevilloside N (5)Amorphous powder; [α]D23 −76.0 (c=0.53, MeOH); IR νmax (film) cm−1: 3346, 2906, 1775, 1715, 1677, 1509, 1214, 1072, 1047; UV λmax (MeOH) nm (log ε): 279 (3.66), 213 (4.42); 1H-NMR (400 MHz, CD3OD) δ: 7.06 (1H, d, J=16 Hz, H-7″), 6.91 (2H, J=9 Hz, H-2, 6), 6.68 (2H, d, J=9 Hz, H-3, 6), 6.49 (1H, dd, J=10, 2 Hz, H-2″), 6.17 (1H, d, J=16 Hz, H-8″), 5.95 (1H, d, J=10 Hz, H-3″), 4.71 (1H, d, J=7 Hz, H-1′), 4.67 (1H, d, J=1 Hz, H-4‴), 4.48 (1H, dd, J=12, 2 Hz, H-6′a), 4.37 (1H, dd, J=12, 6 Hz, H-6′b), 4.02 (1H, td, J=7, 1 Hz, H-5‴), 3.61 (1H, ddd, J=9, 6, 2 Hz, H-5′), 3.58 (2H, d, J=7 Hz, H2-6‴), 3.43 (1H, dd, J=9, 9 Hz, H-3′), 3.41 (1H, dd, J=9, 7 Hz, H-2′), 3.32 (1H, dd, J=9, 9 Hz, H-4′), 3.18 (1H, dd, J=8, 2 Hz, H-6″), 3.09 (1H, d, J=18 Hz, H-5″a), 2.68 (1H, dd, J=18, 8 Hz, H-5″b); 13C-NMR (100 MHz, CD3OD): Table 1; HR-ESI-MS (positive-ion mode) m/z: 633.1430 [M+Na]+ (Calcd for C27H30O16Na: 633.1426).
Grevilloside O (6)Amorphous powder; [α]D27 −15.4 (c=0.59, MeOH); IR νmax (film) cm−1: 3360, 2924, 1712, 1668, 1621, 1511, 1452, 1392, 1282, 1187, 1071; UV λmax (MeOH) nm (log ε): 212 (4.39); 1H-NMR (400 MHz, CD3OD) δ: 6.85 (2H, d, J=10 Hz, H-2″, 6″), 6.68 (1H, d, J=16 Hz, H-7″), 6.65 (1H, d, J=9 Hz, H-5), 6.58 (1H, d, J=3 Hz, H-2), 6.42 (1H, dd, J=9, 3 Hz, H-6), 6.27 (1H, d, J=16 Hz, H-8″), 6.22 (2H, d, J=10 Hz, H-3″, 5″), 4.70 (1H, d, J=7 Hz, H-1′), 4.49 (1H, dd, J=12, 2 Hz, H-6′a), 4.32 (1H, dd, J=12, 6 Hz, H-6′b), 3.60 (1H, ddd, J=8, 6, 2 Hz, H-5′), 3.43 (1H, dd, J=9, 8 Hz, H-3′), 3.38 (1H, dd, J=9, 7 Hz, H-2′), 3.34 (1H, m, H-4′); 13C-NMR (100 MHz, CD3OD): Table 1; HR-ESI-MS (positive-ion mode) m/z: 473.1059 [M+Na]+ (Calcd for C21H22O11Na: 473.1054).
Grevilloside P (7)Amorphous powder; [α]D25 −47.1 (c=0.34, MeOH); IR νmax (film) cm−1: 3357, 2923, 1510, 1219, 1064, 1052; UV λmax (MeOH) nm (log ε): 285 (3.41), 222 (4.01); 1H-NMR (400 MHz, CD3OD) δ: (6.97 (2H, d, J=9 Hz, H-2, 6), 6.71 (2H, d, J=9 Hz, H-3, 5), 4.91 (1H, d, J=7 Hz, H-1′), 4.67 (1H, d, J=2 Hz, H-1‴), 4.62 (1H, d, J=8 Hz, H-1″), 3.88 (1H, dd, J=12, 2 Hz, H-6″a), 3.81 (1H, dd, J=3, 2 Hz, H-2‴), 3.70 (1H, dd, J=12, 5 Hz, H-6″b), 3.67 (1H, dd, J=11, 2 Hz, H-6′a), 3.67 (1H, dd, J=8, 3 Hz, H-3‴), 3.63 (1H, dd, J=8, 7 Hz, H-2′), 3.58 (1H, dq, J=9, 6 Hz, H-5‴), 3.51 (1H, dd, J=11, 5 Hz, H-6′b), 3.41 (1H, dd, J=8, 8 Hz, H-4″), 3.37 (4H, m, H-3′, 5′, 3″, 5″), 3.34 (1H, m, H-4‴), 3.33 (1H, m, H-4′), 3.25 (1H, dd, J=8, 8 Hz, H-2″), 1.23 (3H, d, J=6 Hz, H3-6‴); 13C-NMR (100 MHz, CD3OD): Table 1; HR-ESI-MS (positive-ion mode) m/z: 603.1900 [M+Na]+ (Calcd for C24H36O16Na: 603.1896).
Grevilloside Q (8)Amorphous powder; [α]D23 +16.3 (c=0.43, MeOH); IR νmax (film) cm−1: 3370, 2926, 1710, 1667, 1512, 1313, 1274, 1054; UV λmax (MeOH) nm (log ε): 219 (4.22); 1H-NMR (400 MHz, CD3OD) δ: 6.83 (2H, d, J=10 Hz, H-2″, 6″), 6.69 (1H, d, J=16 Hz, H-7″), 6.25 (1H, d, J=16 Hz, H-8,″ 6.21 (2H, d, J=10 Hz, H-3″, 5″), 5.08 (1/2 H, d, J=4 Hz, H-1′α), 4.48 (1/2 H, d, J=8 Hz, H-1′β), 4.48 (1/2 H, dd, J=12, 2 Hz, H-6′aα), 4.43 (1/2 H, dd, J=12, 2 Hz, H-6′aβ), 4.28 (1/2 H, dd, J=12, 6 Hz, H-6′bα), 4.25 (1/2 H, dd, J=12, 6 Hz, H-6′bβ), 4.00 (1/2 H, ddd, J=10, 6, 2 Hz, H-5α), 3.67 (1/2 H, dd J=9, 9 Hz, H-3′α), 3.51 (1/2 H, ddd, J=9, 6, 2 Hz, H-5′β), 3.37 (1/2 H, dd, J=9, 4 Hz, H-2′α), 3.32 (1/2 H, m, H-3′β), 3.14 (1/2 H, dd, J=9, 8 Hz, H-2′β), H-4′α, H-4′β signals overlapped by the HDO signal; 13C-NMR (100 MHz, CD3OD): Table 1; HR-ESI-MS (positive-ion mode) m/z: 365.0859 [M+Na]+ (Calcd for C15H18O9Na: 365.0843).
Sugar AnalysisAbout 500 µg of each compound (1–8) was hydrolyzed with 1 M HCl (1.0 mL) at 90°C for 2 h. The reaction mixtures were partitioned with an equal amount of EtOAc (1.0 mL), and the water layers were analyzed by HPLC with a chiral detector (JASCO OR-2090plus) on an amino column [Asahipak NH2P-50 4E, Φ=4.6 mm, L=25 cm, CH3CN–H2O (3 : 1), 1 mL/min]. The hydrolyzates of (1–6, 8) each gave a peak for D-glucose at 14.2 min with a positive optical rotation sign. The hydrolyzate of 7 gave peaks for L-rhamnose and D-glucose at 6.8 min and 14.2 min with negative and positive optical rotation signs. The peaks were identified by co-chromatography with authentic L-rhamnose and D-glucose.
Absolute Configuration of 2-Methylbutyric Acid in Grevilloside J (1)Grevilloside J (1) (1.7 mg) and (S)-(+)-2-methylbutyric acid (50 µL) were dissolved in 1 mL of a 1 : 1 mixture of 10% KOH in H2O and 50% aqueous dioxane, and then heated for 1 h at 40°C. The cooled reaction mixtures were neutralized with Amberlite IR-120B (H+) and then the filtrates were evaporated. The residues were analyzed by HPLC (column: Inertsil ODS-3, 6 mm×250 mm; solvent: 20% CH3CN in H2O containing 0.5% trifluoroacetic acid; flow rate: 1.6 mL/min) with a chiral detector (JASCO OR-2090plus) to give a peak of (S)-(+)-2-methylbutyric acid at 17.0 min with a positive optical rotation sign.
Acetylation of Grevilloside P (7)Grevilloside P (7) (5.9 mg) was acetylated with acetic anhydride (500 µL) and pyridine (500 µL) at 25°C overnight. Evaporation of the reagents gave 9.6 mg of grevilloside Q decaacetate (7a). Grevilloside Q decaacetate (7a) Amorphous powder; [α]D21 −38.5 (c=0.20, MeOH); IR νmax (film) cm−1: 2960, 2932, 1752, 1682, 1649, 1247, 1222, 1075, 1046; UV λmax (MeOH) nm (log ε): 267 (3.18), 220 (3.91); 1H-NMR (400 MHz, CD3OD) δ: 7.15 (2H, d, J=9 Hz, H-2, 6), 7.03 (2H, d, J=9 Hz, H-3, 5), 5.35 (1H, d, J=2 Hz, H-1‴), 5.33 (1H, dd, J=9, 3 Hz, H-3‴), 5.32 (1H, dd, J=3, 2 Hz, H-2‴), 5.25 (1H, dd, J=9, 9 Hz, H-3″), 5.20 (1H, m, H-3′), 5.19 (1H, d, J=8 Hz, H-1′), 4.99 (1H, dd, J=9, 9 Hz, H-4″), 4.98 (1H, dd, J=9, 9 Hz, H-4‴), 4.93 (1H, d, J=8 Hz, H-1″), 4.90 (1H, dd, J=10, 9 Hz, H-4′), 4.85 (1H, dd, J=9, 8 Hz, H-2″), 4.33 (1H, dd, J=12, 5 Hz, H-6″a), 4.10 (1H, dd, J=12, 2 Hz, H-6″b), 4.02 (1H, ddd, J=9, 5, 2 Hz, H-5″), 3.99 (1H, ddd, J=10, 7, 2 Hz, H-5′), 3.93 (1H, dq, J=9, 6 Hz, H-5‴), 3.90 (1H, dd, J=9, 8 Hz, H-2′), 3.72 (1H, dd, J=13, 7 Hz, H-6′a), 3.63 (1H, dd, J=13, 2 Hz, H-6′b), 2.25 (6H, s, CH3CO– ×2), 2.15 (3H, s, CH3CO–), 2.03–1.93 (21H, m, CH3CO– ×7), 1.20 (3H, d, J=6 Hz, H3-6‴); 13C-NMR (100 MHz, CD3OD) δ: 172.32, 171.91, 171.88, 171.86, 171.81, 171.71, 171.50, 171.49, 171.47, 171.36 (CH3CO– ×10), 156.1 (C-1), 147.3 (C-4), 123.5 (C-3, 5), 118.5 (C-2, 6), 101.7 (C-1″), 99.9 (C-1′), 99.1 (C-1‴), 78.7 (C-2′), 76.0 (C-3‴), 75.8 (C-5′), 74.4 (C-3″), 73.0 (C-2″), 72.9 (C-5″), 72.4 (C-4‴), 70.7 (C-2‴), 70.6 (C-3′), 70.4 (C-4′), 70.1 (C-4″), 67.9 (C-5‴), 66.5 (C-6′), 63.3 (C-6″), 21.04, 20.92, 20.85, 20.74, 20.72, 20.71, 20.68, 20.65, 20.64, 20.54 (CH3CO– ×10), 18.0 (C-6‴); HR-ESI-MS (positive-ion mode) m/z : 1023.2958 [M+Na]+ (Calcd for C44H56O26Na : 1023.2952).
Tyrosinase Inhibitory ActivityTo the sample solution (20 µL) and 80 µL of 2.5 mM L-dihydroxyphenylalanine solution in 0.1 M phosphate buffer (pH 6.80) 100 µL of mushroom tyrosinase (100 U/mL) in the same buffer was added followed by incubation at 25°C for 5 min. The amounts of dopachrom, formed, were photometrically determined by measuring the optical density (OD) at 405 nm using a microplate reader. Kojic acid was used as a positive control. The inhibition of tyrosinase was calculated as:
Acontrol: without test sample. Ablank: without test sample and tyrosinase.
Cloning of High Melanin Pigment Producing CellsMouse melanoma cells (B16, TKG 0144) provided by the Cell Resource Center for Biomedical Research, Institute of Development, Aging and Cancer, Tohoku University were incubated in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% heat inactivated fetal calf serum supplemented with 100 µg/mL kanamycin (Wako Pure Chemical Industries, Ltd. (Osaka, Japan)) and 5.6 µg/mL of amphotericin B (Sigma) at 37°C under a 5% CO2 atmosphere. From the above mentioned culture, a high melanin-pigment producing cell was cloned by the limiting dilution method and the cloned cell was named B16Y24.
Melanogenesis Inhibitory ActivityB16 and B16Y24 cells (5×103 cells/well) in 400 µL medium were cultured in a 24-multiwell plate containing 40 µL aliquots of sample solutions and 40 µL of 12 mM theophylline solution in DMEM,11) and then the plate was incubated at 37°C under a 5% CO2 atmosphere for 72 h. After removal of the medium, 120 µL of 1 M NaOH was added and the hermetically sealed multiwell plate was stood for 24 h at 37°C. The alkaline solutions (100 µL) were transferred to a 96-well plate and the absorbance of each well was measured at 405 nm using a microplate reader. Arbutin was used as a positive control. The inhibition of melanogenesis was calculated as:
Acontrol: without test sample. Ablank: without test sample and theophylline.
Cytotoxic ActivityCytotoxic activity toward B16 and B16Y24 cells was determined by colorimetric cell viability assay using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT).13) B16 and B16Y24 cells (5×103 cells/well) in 100 µL of DMEM were cultured in the above mentioned medium in a 96-well plate, a 10 µL aliquot of a sample solution, 10 µL of a 12 mM theophylline solution in DMEM were added to each well, and then the plate was incubated at 37°C under a 5% CO2 atmosphere for 72 h. After removal of the medium, a solution of MTT (100 µL, 0.5 mg/mL) was added to each well and the incubation was continued for a further 1 h. The absorbance of each well was measured at 540 nm using a microplate reader. The cytotoxic activity was calculated as:
Acontrol: without test sample. Ablank: without test sample and theophylline.