Results and Discussion
ChemistryPreviously, preparation of the 1,3,4-thiadiazol-2-yl urea derivatives has been reported. The synthesis was accomplished by the reaction of heterocyclic amino compounds with heterocyclic isocyanato compounds in the presence of anhydrous solvent.24,25) In our study, we found that the heterocyclic isocyanato compounds must be freshly made because these compounds were unstable and the reaction was very sensitive to water. To get the 1,3,4-thiadiazol-2-yl urea derivatives in a convenient way, we used a facile one-pot reaction without separation. The synthetic route to the target compounds 6a–k starting from commercially available substituted benzoic acid and thiosemicarbazide was shown in Fig. 1. 5-Substituted-1,3,4-thiadiazol-2-amine 3 was prepared in good yield by stirring mixture of substituted benzoic acid 1, thiosemicarbazide (2) and POCl3 under 80°C for 2 h. Target compounds 6a–k were obtained via a facile one-pot reaction of substituted benzoic acid 4 with diphenylphosphoryl azide (DPPA) under presence of triethylamine to give aromatic isocyanate 5, followed by addition of 5-substituted-1,3,4-thiadiazol-2-amine 3. All the new compounds were fully characterized by spectroscopic techniques (high-resolution mass spectrometry, H-NMR).

Fig. 1. Structure of Rivastigmine and 6a–k
All considered compounds have been assessed as the AChE inhibitors. Their inhibitory potency was described as the half of maximal inhibitory concentration, IC50. The modified Ellman’s method has been used in the evaluation, using the Amplite™ Fluorimetric Acetylcholinesterase Assay Kit.26) The fluorescence intensity of Thiolite™ Green was used to measure AChE activity. For comparative purposes, galanthamine was used as reference compound Table 1. The results were summarized in Table 1.
Table 1. AChE Inhibitory Activity of Target Compounds
6a–k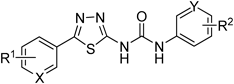 |
|---|
| Compound | X | R1 | Y | R2 | IC50 (µm) |
|---|
| 6a | C | 4-OMe | C | 3,4-Dimethyl | 124.42±7.11 |
| 6b | C | 4-OMe | N | H | 1.17±0.06 |
| 6c | C | 4-OMe | C | 4-OMe | 3.00±0.23 |
| 6d | C | 3,4-Dimethyl | C | 4-OMe | 206.15±12.57 |
| 6e | C | 3,4-Dimethyl | N | H | 111.09±6.34 |
| 6f | C | 3,4-Dimethyl | C | 3,4-Dimethyl | 293.89±14.78 |
| 6g | N | H | C | 4-OMe | 131.31±9.27 |
| 6h | N | H | C | 3,4-Dimethyl | 152.76±11.59 |
| 6i | N | H | N | H | 2.05±0.17 |
| 6j | C | H | C | H | 169.97±1.89 |
| 6k | C | 4-F | C | 4-F | 9.87±0.11 |
| Galanthamine | — | — | — | — | 1.07±0.08 |
The data of AChE inhibition assay clearly showed that most of the designed compounds exhibited high to moderate inhibitory activities. The IC50 values for AChE ranged from 293 to 1.17 µm. In all test compounds, compound 6b (IC50=1.17 µm) had the strongest inhibitory activity of anticholinesterase and was equivalent to reference drug galanthamine (IC50=1.07 µm). The result of biological assay indicated that 1,3,4-thiadiazol-2-yl urea may be a practical and promising skeleton for AChE inhibitors, and would be employed broadly in the design of AChE inhibitor in future.
A simple structure–activity relationship (SAR) analysis showed that AChE inhibition potency was closely related to the substitution of the 5-position of the 1,3,4-thiadiazole core (left aromatic ring) and the substitution of the 1-position of the urea group (right aromatic ring). Comparing four groups of synthesized compounds (bioactivity sequence: 6a>6h>6f, 6b>6i>6e, 6c>6g>6d, 6k>6j>6f), it seemed that compounds in which left aromatic ring was electron-deficient, such as pyridine, or was substituted by electron-withdrawing group, such as fluorine, in general gave better AChE inhibition activity. Substitution with methoxy goup was an exception, which was an electron-donating group, but also promoted AChE inhibition activity. Furthermore, it could be found that methoxy group substitution had more contribution than pyridine and fluorine substitution in the left hand. SAR analysis concerning of right aromatic ring could also help us to discover the similar conclusion. The effect of electron-deficiency of the aromatic rings could be further confirmed by comparison of AChE inhibitory actives of 6k, 6j and 6f. Substitution with fluorine (IC50 value of 6k was 9.87 µm) was obviously beneficial to AChE inhibitory activity. Concerning methoxy group in right and left hand, although it acted as an electron-donating group by its mesomeric effect, it also enhanced the AChE inhibitory activity, for instance, compound 6c. The oxygen of methoxy group, which was a hydrogen acceptor, may play an important role in AChE inhibition, but understanding the exact reason for the difference of inhibitory activities need further study. In our lab, we are trying to prepare co-crystal of AchE and compound 6b, which is most potential in our compounds. X-Ray crystallographic analysis and further pharmacological study, such as in vivo evaluation, will be reported in future.
Experimental
All reagents were purchased from Shanghai Chemical Reagent Company. Column chromatography (CC): silica gel 60 (200–300 mesh). Thin-layer chromatography (TLC): silica gel 60 F254 plates (250 mm; Qingdao Ocean Chemical Company, China). mp: capillary tube; uncorrected. 1H-NMR spectra: Bruker ACF-300Q apparatus at 300 MHz, in DMSO-d6 unless otherwise indicated δ; δ in ppm rel. to Me4Si, J in Hz. High resolution-time-of-flight (HR-TOF)-MS were measured on a BioTOFTM-Q mass spectrometer (Bruker); in m/z.
General Procedure for Preparing Compounds 3a–kA mixture of substituted benzoic acid (40.61 mmol) and thiosemicarbazide (48.73 mmol) in POCl3 (45 mL) was stirring for 2 h at 80°C. After cooling to room temperature, water (60 mL) was added in 30 min. Ice-water was added, and the pH was adjusted to 8 with 1 m sodium hydroxide solution. The mixture was filtered. The obtained solid was crystallized from DMF–H2O to give 3a–k.
General Procedure for Preparing Compounds 6a–kA mixture of substituted benzoic acid (3 mmol) and triethylamine (3.3 mmol) in anhydrous toluene 40 mL was stirring for 45 min at room temperature. To the above reaction mixture DPPA (3.6 mmol) was added and heated to 40°C for 1 h. Then the reaction mixture was refluxed for 3 h. After cooling to room temperature, intermediate 3a–k (3 mmol) was added and refluxed for 10 h. Cooled to room temperature and concentrated to give the crude product. The crude product was refluxed in 50 mL methanol for 0.5 h. After cooling, the mixture was filtered and the solid was collected as target product.
1-(3,4-Dimethylphenyl)-3-(5-(4-methoxyphenyl)-1,3,4-thiadiazol-2-yl)urea (6a): Yield 75.3%; mp 317.1–319.4°C. 1H-NMR (300 MHz, DMSO-d6) δ: 2.17–2.20 (d, 6H), 3.82 (s, 3H), 7.06–7.84 (m, 7H), 8.81 (s, 1H), 10.88 (s, 1H). HR-TOF-MS m/z 355.1227 ([M+H]+, C18H19N4O2S+; Calcd 355.1223).
1-(5-(4-Methoxyphenyl)-1,3,4-thiadiazol-2-yl)-3-(pyridine-3-yl)urea (6b): Yield 53.0%; mp 317.6–320.1°C. 1H-NMR (300 MHz, DMSO-d6) δ: 3.72 (s, 3H), 7.07–7.10 (d, J=9 Hz, 2H), 7.83–7.86 (d, J=9 Hz, 2H), 7.34–7.38 (m, 1H), 7.97–8.00 (d, J=9 Hz, 1H), 8.26–8.27 (d, J=3 Hz, 1H), 8.69 (s, 1H), 9.291 (s, 1H). HR-TOF-MS m/z 328.0866 ([M+H]+, C15H14N2O2S+; Calcd 328.0863).
1-(5-(4-Methoxyphenyl)-1,3,4-thiadiazol-2-yl)-3-(4-methoxyphenyl)urea (6c): Yield 85.7%; mp 314.0–316.9°C. 1H-NMR (300 MHz, DMSO-d6) δ: 3.03 (s, 3H), 3.82 (s, 3H), 6.89–6.92 (d, J=9 Hz, 2H), 7.06–7.09 (d, J=9 Hz, 2H), 7.39–7.42 (d, J=9 Hz, 2H), 7.82–7.85 (d, J=9 Hz, 2H), 8.86 (s, 1H), 10.9 (s, 1H). HR-TOF-MS m/z 357.1020 ([M+H]+, C17H17N4O3S+; Calcd 357.1016).
1-(3,4-Dimethylphenyl)-3-(5-(4-methoxyphenyl)-1,3,4-thiadiazol-2-yl)urea (6d): Yield 76.4%; mp 329.4–331.7°C. 1H-NMR (300 MHz, DMSO-d6) δ: 2.28–2.31 (d, 6H), 3.74 (s, 3H), 6.89–6.93 (d, J=9 Hz, 2H), 7.39–7.42 (d, J=9 Hz, 2H), 7.27–7.29 (d, J=6 Hz, 1H), 7.60–7.62 (d, J=6 Hz, 1H), 7.68 (s, 1H), 8.86 (s, 1H), 10.90 (s, 1H). HR-TOF-MS m/z 355.1230 ([M+H]+, C18H19N4O2S+; Calcd 355.1223).
1-(5-(3,4-Dimethylphenyl)-1,3,4-thiadiazol-2-yl)-3-(pyridine-3-yl)urea (6e): Yield 60.7%; mp 327.4–330.6°C. 1H-NMR (300 MHz, DMSO-d6) δ: 2.28 (s, 3H), 2.30 (s, 3H), 7.27–7.29 (d, J=6 Hz, 1H), 7.61–7.63 (d, J=6 Hz, 1H), 7.68 (s, 1H), 7.34–7.38 (m, 1H), 7.97–8.00 (d, J=9 Hz, 1H), 8.26–8.27 (d, J=3 Hz, 1H), 8.70 (s, 1H), 9.30 (s, 1H), 11.28 (s, 1H). HR-TOF-MS m/z 326.1074 ([M+H]+, C16H16N5OS+; Calcd 326.1070).
1-(3,4-Dimethylphenyl)-3-(5-(3,4-dimethylphenyl)-1,3,4-thiadiazol-2-yl)urea (6f): Yield 81.9%; mp 341.2–344.6°C. 1H-NMR (300 MHz, DMSO-d6) δ: 2.17–2.20 (d, 6H), 2.27–2.29 (d, 6H), 7.06–7.08 (d, J=6 Hz, 1H), 7.20–7.29 (m, 3H), 7.60–7.62 (d, J=6 Hz, 1H), 7.68 (s, 1H), 8.86 (s, 1H), 10.92 (s, 1H). HR-TOF-MS m/z 353.1436 ([M+H]+, C19H21N4OS+; Calcd 353.1431).
1-(4-Methoxyphenyl)-3-(5-(pyridine-3-yl)-1,3,4-thiadiazol-2-yl)urea (6g): Yield 50.8%; mp 321.6–324.4°C. 1H-NMR (300 MHz, DMSO-d6) δ: 3.75 (s, 3H), 6.92–7.94 (d, J=6 Hz, 2H), 7.42–7.44 (d, J=6 Hz, 2H), 7.56–7.59 (m, 1H), 8.29–8.31 (d, J=6 Hz, 1H), 8.70–8.71 (d, J=3 Hz, 1H), 8.92 (s, 1H), 9.11 (s, 1H), 11.13 (s, 1H). HR-TOF-MS m/z 328.0868 ([M+H]+, C15H14N5O2S+; Calcd 328.0863).
1-(3,4-Dimethylphenyl)-3-(5-(pyridine-3-yl)-1,3,4-thiadiazol-2-yl)urea (6h): Yield 58.9%; mp 318.2–321.6°C. 1H-NMR (300 MHz, DMSO-d6) δ: 2.05–2.31 (m, 6H), 7.08–7.29 (m, 3H), 7.56 (s, 1H), 8.29–8.30 (m, 1H), 8.70 (s, 1H), 8.90 (s, 1H), 9.10 (s, 1H), 11.16 (s, 1H). HR-TOF-MS m/z 326.1076 ([M+H]+, C16H16N5OS+; Calcd 326.1070).
1-(Pyridine-3-yl)-3-(5-(pyridine-3-yl)-1,3,4-thiadiazol-2-yl)urea (6i): Yield 36.0%; mp 319.4–322.4°C. 1H-NMR (300 MHz, DMSO-d6) δ: 7.34 (m, 1H), 7.57–7.58 (m, 1H), 7.99–8.00 (d, J=3 Hz, 1H), 8.29–8.31 (d, J=6 Hz, 2H), 8.70 (s, 2H), 9.10 (s, 1H), 9.34 (s, 1H), 11.78 (s, 1H). HR-TOF-MS m/z 299.0714 ([M+H]+, C13H11N6OS+; Calcd 299.0710).
1-(5-Phenyl-1,3,4-thiadiazol-2-yl)-3-phenyl Urea (6j): Yield 75.8%; mp 309.5–311.9°C. 1H-NMR (300 MHz, DMSO-d6) δ: 7.04–7.09 (m, 1H), 7.31–7.36 (m, 2H), 7.89–7.91 (m, 2H), 7.50–7.52 (m, 5H), 9.05 (s, 1H), 11.04 (s, 1H). HR-TOF-MS m/z 295.0660 ([M−H]−, C15H11N4OS−; Calcd 295.0654).
1-(5-(4-Fluorophenyl)-1,3,4-thiadiazol-2-yl)-3-(4-fluorophenyl)urea (6k): Yield 81.2%; mp 335.7–336.9°C. 1H-NMR (300 MHz, DMSO-d6) δ: 7.14–7.20 (t, J=8.7 Hz, 2H), 7.34–7.40 (t, J=8.7 Hz, 2H), 7.51–7.55 (t, J=8.1 Hz, 2H), 7.94–7.99 (t, J=8.4 Hz, 2H), 9.13 (s, 1H), 11.14 (s, 1H). HR-TOF-MS m/z 331.0472 ([M−H]−, C15H9N4OSF2−; Calcd 331.0465).
In Vitro AChE Inhibition AssayAChE inhibitory activity was detected by a microtitre plate assay based on Ellman’s method26) (Rhee et al., 2001), using the Amplite™ Fluorimetric Acetylcholinesterase Assay Kit. In 96-well plates, 25 µL Acetylcholine, 2 µL Thiolite™ Green, 62.2 µL of buffer and 10 µL of the test compound at concentration of 0.01, 0.1, 1, 10, 100, 1000 µm, 10 mm were added and the fluorescence intensity was measured at 485/528 nm every 13 s for five times. After adding 25 µL of 0.125 U/mL AChE, the fluorescence intensity was read again every 13 s for five times. The fluorescence intensity was measured using a Multi-Plate Reader (Synergy HT, BioTek Ltd.). Percentage of inhibition was calculated by comparing the rates for the sample to the blank (DMSO), control contained all components except the tested extract. Galanthamine was used as positive control. The mean of four measurements for each concentration was determined (n=4). The 50% inhibitory concentration (IC50) was calculated from a doseeresponse curve obtained by plotting the percentage of inhibition versus the log concentration with the use of GraphPad Prism 5.0 software.

