Regular Articles
Preparation and Evaluation of Solid Dispersion of Atorvastatin Calcium with Soluplus® by Spray Drying Technique
2014 Volume 62 Issue 6 Pages 545-551
Details
2014 Volume 62 Issue 6 Pages 545-551
The aim of the present study was to investigate the effect of Soluplus® on the solubility of atorvastatin calcium and to develop a solid dispersion formulation that can improve the oral bioavailability of atorvastatin calcium. We demonstrated that Soluplus® increases the aqueous solubility of atorvastatin calcium. Several solid dispersion formulations of atorvastatin calcium with Soluplus® were prepared at various drug : carrier ratios by spray drying. Physicochemical analysis demonstrated that atorvastatin calcium is amorphous in each solid dispersion, and the 2 : 8 drug : carrier ratio provided the highest degree of sustained atorvastatin supersaturation. Pharmacokinetic analysis in rats revealed that the 2 : 8 dispersion significantly improved the oral bioavailability of atorvastatin. This study demonstrates that spray-dried Soluplus® solid dispersions can be an effective method for achieving higher atorvastatin plasma levels.
Solid dispersion formulations that combine one or more active pharmaceutical ingredients (API) with an inert carrier polymer or polymer matrix are frequently developed to improve the dissolution or solubility characteristics of an API.1) Solid dispersions are prepared by solvent, melting, or solvent-melting methods, and the properties of the resulting dispersion are influenced by the physicochemical properties of both the API and the carrier. Upon dissolution of an amorphous solid dispersion, API molecules are dispersed molecularly with the carrier polymer, stabilizing the amorphous form of the API. In many cases, this improves the solubility and dissolution of the API compared to the crystalline material, since an amorphous form may lead to higher levels of supersaturation.2,3) Consequently, amorphous solid dispersion has become an increasingly utilized approach for improving the bioavailability of poorly water-soluble compounds.4–7)
In many cases, a supersaturated drug solution generated by amorphous solid dispersion is thermodynamically unstable and energetically tends to return to equilibrium via precipitation.8) To maintain supersaturation, precipitation must be inhibited, in many cases by a polymeric precipitation inhibitor.9) Several commercially available polymeric precipitation inhibitors have been explored in this context, including polyvinyl pyrrolidone (PVP), polyethylene glycol (PEG), and hydroxypropyl methyl cellulose (HPMC).10) However, the selection of an appropriate polymeric precipitation inhibitor is still largely empirical, and reliable structure–activity relationships have not been established.11) In this study, a relatively new amphiphilic polymer, Soluplus®, was investigated as a carrier polymer and precipitation inhibitor for making atorvastatin calcium solid dispersions. Soluplus® is a polyvinyl caprolactam–polyvinyl acetate–polyethylene glycol graft copolymer (13% PEG 6000/57% vinyl caprolactam/30% vinyl acetate), shows excellent solubilizing properties for poorly water-soluble APIs, and offers the possibility of generating amorphous solid dispersions by hot-melt extrusion.12)
The aim of the present study was to investigate the effect of Soluplus® on the solubility of atorvastatin calcium and to develop an effective solid dispersion formulation that improves the oral bioavailability of atorvastatin calcium. Atorvastatin calcium is a Biopharmaceutics Classification System (BCS) class II drug that is insoluble in aqueous solutions of pH 4 and very slightly soluble in distilled water or pH 7.4 phosphate buffer.13) Atorvastatin calcium has an absolute bioavailability (% F) of 12% from a 40 mg oral dosage form.14) In this study, solid dispersion formulations of atorvastatin calcium with several drug/Soluplus® ratios were prepared by spray drying, and the dissolution profiles of the resulting dispersions were compared to those of crystalline atorvastatin calcium, and amorphous atorvastatin calcium. The oral bioavailability of solid dispersion was evaluated in rats.
Atorvastatin calcium trihydrate (Form I) was obtained from Zhejiang Jiangbei Pharmaceutical Co., Ltd. (China). Soluplus® was kindly supplied by BASF (BASF, Ludwigshafen, Germany). Metaqualone was purchased from Sigma (St. Louis, MO, U.S.A.). All organic solvents were of high-performance liquid chromatography (HPLC) grade. All other chemicals were of reagent grade.
Phase Solubility AnalysisPhase solubility analysis was carried out by adding excess amounts (50 mg) of atorvastatin calcium into a capped glass vial containing 10 mL of water with varying Soluplus® concentrations (0–10 mg/mL). Samples were sonicated for 30 min at room temperature and incubated in a shaking water bath (60 rpm) at 37±0.1°C for 48 h. After incubation, an aliquot of the sample was immediately filtered through a 0.22-µm nylon syringe filter (Whatman Inc., U.S.A.). The filtrate was diluted with methanol, and the concentration of atorvastatin was determined by HPLC analysis using a C18 analytical column (Xterra, 5 µm, 4.6 mm×250 mm, Waters), an LC 10ADvp pump, and an SPD-10ADvp UV detector monitoring absorbance at 245 nm (Shimadzu, Japan). The mobile phase was a 60 : 40 mixture of acetonitrile and 50 mm sodium acetate in water, adjusted to pH 4.0 with glacial acetic acid. The injection volume was 20 µL and the eluent flow rate was 1.0 mL/min.
Preparation of Solid DispersionsSpray-dried solid dispersions of atorvastatin calcium and Soluplus® at 5 : 5, 4 : 6, 3 : 7, and 2 : 8 weight ratios were prepared by initial dissolution in methanol at a concentration of 1% w/v of solute followed by spray drying with a laboratory-scale spray dryer (SD 1000, Eyela, Japan) under the following conditions: 65–80°C inlet temperature, 50–60°C outlet temperature, 2–3 mL/min feed rate, 10 kPa atomization air pressure, and 0.60–0.70 m3/min drying air flow rate. The amorphous atorvastatin calcium microparticles were also prepared by spray drying at same condition, based on previously reported method.15) As a control, a non-spray dried physical mixture was also prepared by mixing atorvastatin with Soluplus® at a 2 : 8 ratio with a mortar and pestle until homogeneous. All samples were passed through a 250-µm sieve (mesh size 60) and stored in a desiccator at room temperature prior to analysis.
Characterization of Solid DispersionsDrug content was measured by HPLC analysis of weighed aliquots of spray-dried solid dispersions of atorvastatin following dissolution in methanol and sonication for 10 min. Morphology of the particles was examined by scanning electron microscopy (SEM; JSM-7100f; Jeol Ltd., Japan). The particle size of spray-dried solid dispersion particles was determined using a HELOS laser diffraction analyzer (Sympatec GmbH, Germany) equipped with a RODOS vibrating trough disperser. The specific surface area of the samples was determined by N2 adsorption using Surface Area Analyzer ASAP 2010 (Micromeritics Instrument Corporation, U.S.A.). X-Ray diffraction patterns were recorded using a D8 Advance X-ray diffraction system (Bruker AXS GmbH, Germany) with Ni-filtered CuKα radiation. The 2θ scan range was 5–60° with a step size of 0.02° at a rate of 3°/min. FT-IR spectra were collected on a FT-IR spectrometer (Spectrum GX, PerkinElmer, Inc., U.S.A.) using attenuated total reflectance (ATR) method. The scanning range was 650–4000 cm−1.
Supersaturated Dissolution AnalysisSupersaturated dissolution tests were performed using a USP rotating paddle apparatus with a VK 7000 dissolution testing station and a VK 750d heater/circulator (Vankel, U.S.A.). Tests were conducted in accurately weighed samples containing the 300 mg of atorvastatin calcium were dispersed in 300 mL of water and incubated at 37°C with shaking at 100 rpm, and 2-mL aliquots were collected at different time intervals, filtered through a 0.22-µm nylon syringe filter, and analyzed for atorvastatin concentration by HPLC.
Measuring the Inhibitory Effect of Soluplus® on Atorvastatin RecrystallizationAtorvastatin calcium was dissolved in methanol at 300 mg/mL. One-milliliter aliquots of the atorvastatin solution were added to 300-mL water samples with different concentrations of Soluplus® (0–1 mg/mL) in a United States Pharmacopeia (USP) rotating paddle apparatus (USP apparatus II) at 37±0.1°C and 150 rpm. Samples (2 mL) were removed at specified time points, filtered using a 0.22-µm nylon syringe filter, and analyzed for atorvastatin concentration by HPLC.
In Vivo Animal StudyAll animal protocols were approved by the animal care committee of Kyungsung University according to the guidelines published in the “Guidelines for the Care and Use of Laboratory Animals.” The bioavailability of the solid dispersion was evaluated in Male Sprague-Dawley rats weighing 240–260 g (Samtaco Bio Korea Inc., Korea). The animals were divided into three groups of five animals that each received a 1-mL oral dose of an aqueous suspension containing amorphous atorvastatin, physical mixture, or solid dispersion (atorvastatin : Soluplus® at a 2 : 8 ratio) equivalent to 10 mg/kg of atorvastatin. Serial blood samples (approximately 350 µL each) were collected from the femoral artery at specified time intervals, placed in heparinized tubes, and separated by centrifugation at 10000 rpm for 10 min. The samples were extracted and atorvastatin plasma concentrations determined by LC-MS as reported previously.16) Briefly, 200-µL plasma samples were combined with 700 µL of acetonitrile and 100 µL of internal standard solution (100 ng/mL methaqualone), vortexed for 60 s, and centrifuged at 13000 rpm for 10 min at 1°C, with the supernatants immediately transferred to clean tubes. The organic phase was evaporated to dryness under nitrogen at 40°C for 10 min. The dried residue was reconstituted in 200 µL of methanol, and 5 µL of each sample was analyzed by LC-MS on a system consisting of a Kromasil C18 column (150×4.6 mm, 5 µm), LC-10ADvp pump, SIL-10A auto-injector, SPD-10ADvp UV detector monitoring absorbance at 270 nm, and an LCMS-2010 A mass spectrometer (Shimadzu, Japan). The mobile phase was 0.1 m ammonium acetate buffer (pH 4.0)/acetonitrile (50 : 50, v/v) and the flow rate was 1.0 mL/min. The mass spectrometer was operated in positive-ion mode and was connected to the chromatographic system using an atmospheric pressure ionization (API) electrospray interface. The [M+H]+, m/z 251.00 for methaqualone, and [M+H]+, m/z 559.00 for atorvastatin, were selected as detecting ions. The MS operating conditions were optimized as follows: 1.5 L/min drying gas, 250°C CDL temperature, 200°C block temperature, and +4.5 kV probe voltage. Data processing was performed using the LC-MS solution software (Shimadzu, Japan). The Cmax and Tmax were calculated directly from plasma data, and the area under curve from 0 to 8 h (AUC0→8 h) was calculated using non-compartmental analysis (WinNonlin 2.1; Pharsight Corp., Mountain View, CA, U.S.A.). Pharmacokinetic parameters obtained from different formulations were compared for statistically significant differences (p<0.05) by one-way ANOVA, least significance difference (LSD) test, and the Student–Newman–Keul test using SPSS 20.0 software (IBM Corporation, U.S.A.).
The impact of various Soluplus® concentrations on the solubility of atorvastatin calcium was evaluated with phase solubility techniques. As predicted, increasing the Soluplus® concentration improves the solubility of atorvastatin calcium (Fig. 1). At the highest Soluplus® concentration (10 mg/mL), atorvastatin calcium solubility was increased approximately fourfold compared to its aqueous solubility without Soluplus® (150.7±7.83 µg/mL at 37°C). The phase solubility diagrams for these data would be classified as Type AL curves according to the definition of Higuchi and Connors.17) The Gibbs free energy of transfer (ΔGt°) of atorvastatin calcium from water alone to an aqueous solution with Soluplus® was calculated using the following equation: ΔGt°=−2.303RT log(Sc/S0) where Sc/S0 is the ratio of solubility of atorvastatin calcium with Soluplus® to the solubility without Soluplus®. As shown in Table 1, ΔGt° values were negative, indicating that the transfer of atorvastatin calcium from water alone to an aqueous solution of Soluplus® was spontaneous and energetically favorable. Furthermore, ΔGt° values decreased with increasing Soluplus® concentration, demonstrating that the solubilization process became more favorable with higher Soluplus® concentrations.

| Soluplus® concentration (mg/mL) | ΔGt° (kJ/mol) |
|---|---|
| 0.1 | −0.258 |
| 0.5 | −0.694 |
| 1 | −1.034 |
| 2.5 | −1.779 |
| 5 | −2.523 |
| 10 | −3.597 |
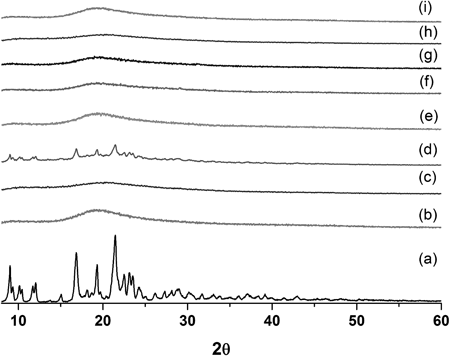
Physicochemical characterization of spray-dried solid dispersions indicated properties typical of drug : polymer dispersions. SEM indicated microparticles that were of a regular spherical shape with shallow dents formed during the rapid evaporation of solvent (Fig. 2). The mean particle size and specific surface areas were 6.55–9.06 µm and 1.15–1.42 m2/g, respectively (Table 2), and there are no significant difference between solid dispersions (p>0.1). In fact, the morphology and particle size of the solid dispersion was not influenced by the drug : carrier ratio. HPLC analysis confirmed that the atorvastatin calcium content for the solid dispersions agreed well with the calculated values. Powder X-ray diffraction (PXRD) analysis of crystalline atorvastatin calcium showed characteristic diffraction peaks at 2θ=9.12°, 9.44°, 16.97°, 19.45°, and 21.59° (Fig. 3). The diffraction patterns for all solid dispersions were markedly different from that of the crystalline form and indicated an amorphous or molecular dispersion state.

| Formulation (drug : Soluplus®) | Drug contenta) | Volume mean particle sizea) (µm) | Specific surface areaa) (m2/g) |
|---|---|---|---|
| Amorphous microparticle | — | 6.71±1.31 (1.80)b) | 1.91±0.05 |
| Solid dispersion 5 : 5 | 97.3±2.1% | 8.54±1.52 (1.80) | 1.42±0.02 |
| Solid dispersion 4 : 6 | 99.1±1.9% | 7.77±1.21 (1.78) | 1.33±0.05 |
| Solid dispersion 3 : 7 | 98.5±1.5% | 6.55±1.01 (1.86) | 1.21±0.06 |
| Solid dispersion 2 : 8 | 97.5±1.8% | 9.06±1.78 (1.78) | 1.15±0.03 |
a) Mean±S.D. n=3. b) SPAN=(d90−d10)/d50, where d10, d50, and d90 are the diameter sizes and the given percentage value is the percentage of particles smaller than that size.
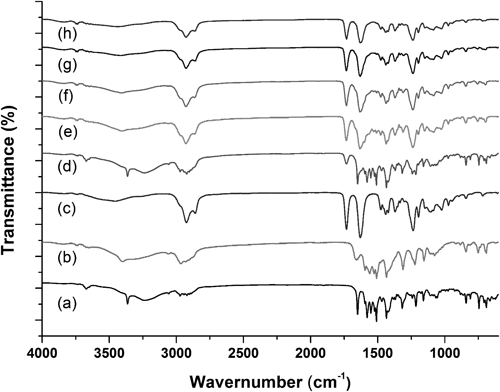
FT-IR spectra of all samples were used to assess possible molecular interactions between atorvastatin calcium and Soluplus® in the solid state (Fig. 4). Characteristic IR peaks for crystalline atorvastatin calcium were observed at 3362 cm−1, 3222 cm−1, 1649 cm−1, and 1578 cm−1 which can be attributed to N–H stretching, asymmetric O–H stretching, asymmetric C=O stretching, and symmetric C=O stretching, respectively, while amorphous atorvastatin calcium exhibited peaks at 3222 cm−1, 1649 cm−1, and 1578 cm−1.16) The spectra of Soluplus® showed peaks at 2924 cm−1 (aliphatic C–H stretching), 1732 cm−1, and 1631 cm−1 (C=O stretching).18,19) FT-IR spectra of a physical mixture of atorvastatin calcium and Soluplus® at a 2 : 8 ratio correspond to a summation of crystalline atorvastatin calcium and Soluplus® spectra, suggesting that no interactions between the two species or disruption of drug crystallinity occur in the mixtures. Notably, however, the spectra of solid dispersions lack peaks characteristics of crystalline atorvastatin calcium and showed peaks representative of Soluplus® and amorphous atorvastatin calcium. These results are consistent with the results of PXRD.
Dissolution analysis demonstrated that all solid dispersions tested rapidly dissolve and yield maximal atorvastatin calcium supersaturation within 4 h (Fig. 5). The maximum concentration attained increased with higher Soluplus® ratios, with the 2 : 8 dispersion (drug : Soluplus®) showing maximal solubility enhancement of 6.6-fold compared to the crystalline form and 2.5-fold compared to the physical mixture. Indeed, dissolution of the 2 : 8 dispersion reached a maximum soluble concentration close to 1 mg/mL (100%) that was maintained for more than 24 h. In comparison, the maximal soluble concentration of amorphous atorvastatin calcium was 3.1-fold higher than that of the crystalline form and decreased with time. This time-dependent decrease is attributable to recrystallization due to water-induced plasticization of the amorphous system.16,20) Thus, the inclusion of Soluplus® both increases the maximal solubility and prevents recrystallization, most likely via blocking the crystallization surface and providing steric stabilization.10,21) This inhibitory effect of Soluplus® on recrystallization was investigated by diluting a concentrated solution of atorvastatin calcium into water with different Soluplus® concentrations and monitoring the atorvastatin calcium concentration over time. As expected, the concentration of atorvastatin calcium rapidly decreased after dilution in water without Soluplus®, to a maximum concentration comparable to that of the amorphous form. Correspondingly, the inclusion of Soluplus® enables a higher atorvastatin calcium concentration to be maintained, up to 890 µg/mL for 24 h when 1 mg/mL Soluplus® is present (Fig. 6). Thus, Soluplus® maintains a high supersaturation of atorvastatin calcium by inhibiting both drug nucleation and crystal growth.
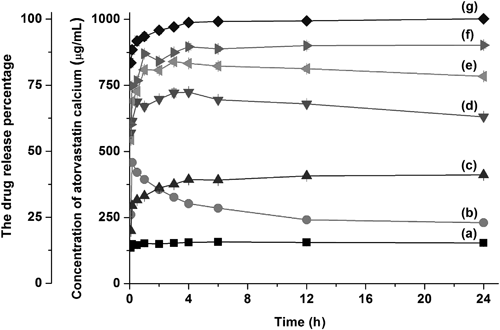
Data shown are mean±standard deviation with n=3.

Data shown are the mean±standard deviation with n=3. The dashed line indicates the solubility of atorvastatin calcium in 1 mg/mL Soluplus® solution.
Based on its maximal enhancement of solubility in aqueous solution, the 2 : 8 solid dispersion formulation was selected for bioavailability evaluation in rats. As predicted, the 2 : 8 solid dispersion showed significantly higher bioavailability than either the physical mixture or amorphous drug (Fig. 7, Table 3), with a maximal atorvastatin plasma concentration (Cmax) that was 3.6- and 2.3-fold higher than that of the physical mixture or amorphous atorvastatin, respectively. It is reasonable to conclude that this bioavailability increase is largely because of enhanced drug concentrations in the gastroinstestinal tract.22) Overall, the significant differences in the plasma concentration–time profiles of atorvastatin between the solid dispersion and amorphous atorvastatin calcium microparticle after oral administration might have been associated with the increased supersaturated concentration over an extended period of time in water. However, in order to better predict the correlation between in vivo supersaturation and in vivo pharmacokinetic behavior, further study on the solubility and dissolution characteristics of the solid dispersions in different media types will be needed. Nevertheless, our results suggest that Soluplus® solid dispersion formulations can improve the supersaturation and oral bioavailability of atorvastatin calcium.
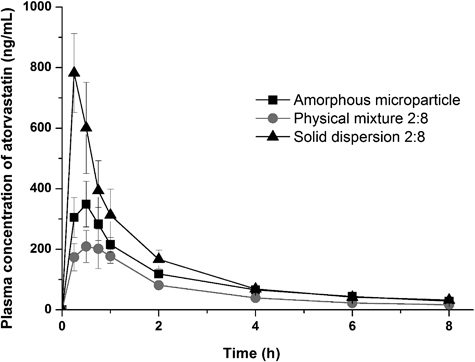
Data shown are mean±standard deviation with n=5.
| Formulation (drug : Soluplus®) | Pharmacokinetic parametera) | ||
|---|---|---|---|
| Cmax (ng/mL) | AUC0→8 h (ng·h/mL) | Tmax (h) | |
| Amorphous microparticle | 348.6±53.9b) | 791.1±116.5b) | 0.54±0.28 |
| Physical mixture 2 : 8 | 215.1±31.0 | 528.9±97.5 | 0.89±0.66 |
| Solid dispersion 2 : 8 | 783.9±124.9b,c) | 1279.6±211.4b,c) | 0.30±0.11 |
a) Mean±S.D. n=5. b) Significant at p<0.05 vs. physical mixture. c) Significant at p<0.05 vs. amorphous microparticle.
In addition, the drug content of 2 : 8 solid dispersion was 97.2±1.0% after one year at 25±2°C and 60±5% relative humidity, indicating no significant chemical degradation (initial: 97.5±1.8%). Furthermore, the X-ray diffraction patterns of the 2 : 8 solid dispersion were similar on day 0 and after one year (Fig. 3). These results indicate that the amorphous state of atorvastatin calcium in solid dispersion remained almost unchanged for one year. In fact, the 2 : 8 solid dispersion formulation in this study is found to be stable at least for one year under the long-term condition.
In this study, the dissolution and bioavailability characteristics were evaluated for several spray-dried solid dispersions of atorvastatin calcium and Soluplus®. In all dispersions, the drug maintained an amorphous state, and the 2 : 8 drug : Soluplus® formulation yielded the maximal solubility enhancement that was sustained for 24 h. Pharmacokinetic analysis in rats indicated that the 2 : 8 dispersion significantly improved the bioavailability of atorvastatin compared to both the amorphous drug and a physical mixture of drug and Soluplus®. Thus, spray-dried solid dispersions using Soluplus® can be an effective method for enhancing the bioavailability of atorvastatin calcium.
This work was supported by Pusan National University Research Grant, 2013.