Results and Discussion
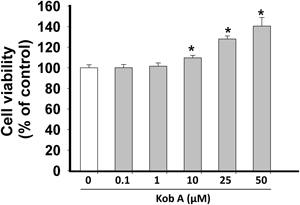
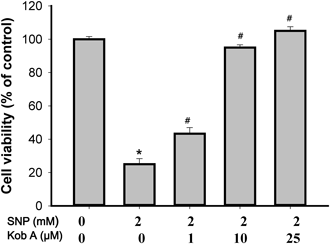
First, we tested the possible cytotoxic effect of Kob A on cardiomyoblast H9c2 cells for 24 h. Although there was no significant cytotoxicity (Fig. 2), Kob A showed a cell proliferative effect at the concentrations of 10 (109.7±2.3%), 25 (128±2.9%), and 50 µM (140.5±8.3%). The exposure of cells to SNP at 1, 2, and 5 mM for 24 h caused a significant decrease in cell viability to 76.1±1.6%, 25.1±3.3%, and 17.3±0.14% as compared to untreated control, respectively (p<0.05). Thus we used 2 mM SNP for inducing cell death and finding a possible underlying mechanism. As shown in Fig. 3, the survival rate of H9c2 cells was significantly augmented in the presence of Kob A as a dose-dependent manner.
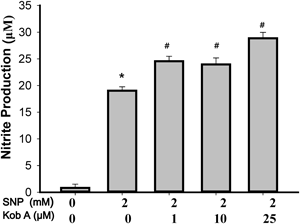
Excessively released NO from SNP can kill the cells but NO scavenging chemicals, such as the carboxyl derivative of 2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide (PTIO), suppress NO- or SNP-mediated cell death.5) To rule out that the cell protection against SNP by Kob A is not a mere reflection of Kob A-mediated NO scavenging property, we then measured the NO production from SNP in the presence or absence of Kob A using Greiss reaction.15) SNP stimulation for 24 h resulted in a remarkably increased NO production (19.0±0.7 µM) when compared to the untreated control (0.8± 0.7 µM). NO production from SNP was not decreased in the presence of Kob A at 24 h but was slightly increased when compared to 2 mM SNP (Fig. 4). This increase of NO production may be related to more NO production from surviving cells.
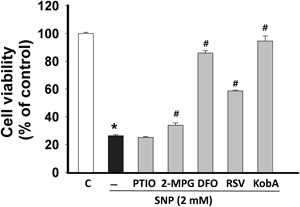
To exclude the potential involvement of NO and to find the direct cellular damaging moiety caused by SNP in H9c2 cells, the effects of deferoxamine (DFO; an iron chelating agent, 50 µM),16) 2-mercaptopropionylglycine (2-MPG; a reactive oxygen species (ROS) scavenger, 300 µM),17) (PTIO; a NO scavenger, 100 µM),18) resveratrol (phytoestrogen with ROS scavenging activity, 50 µM)19) on SNP-triggered H9c2 cells death were tested (Fig. 5). The cell viability of PTIO, DFO, RSV, and 2-MPG treatment without SNP on H9c2 cells were 99.7±0.8, 90.4±1.3, 94.2±0.6, and 108.5±1.2%, respectively (n=12). The effects of PTIO (100 µM), 2-MPG (300 µM), DFO (50 µM), and RSV (50 µM) on SNP-mediated H9c2 cell death were tested by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (Fig. 5). NO-scavenger PTIO did not show any significant protective effect on SNP-mediated cell death (cell viability; 25.2±0.6, p>0.05 vs. SNP). When treating with 2-MPG for scavenging a free radical derived from SNP, there was only a slight increase in cell viability (33.9±1.7, p<0.05). Unlike a previous report in MG-63 cells,20) the protective effects of 2-MPG and PTIO against SNP insults were not found in H9c2 cells. These data may exclude, at least in part, the possible involvement of excessive NO in SNP-mediated death in H9c2 cells. In addition, it is plausible that different cellular states may yield different outcomes in the setting of SNP insults since the toxicity of NO may be closely related to the handling of iron in the cells.16) However, DFO (85.9±3.8%) and RSV (58.6±0.7%) showed a remarkable cellular protection against SNP-mediated toxicity (p<0.05). In this study, RSV showed a cellular protective effect against SNP insults but the protection was weaker than that of Kob A. RSV has been known as a powerful antioxidant but it also has other biological effects.19) The protective potential of RSV and Kob A may not be completely attributed to its antioxidant capacity, since the two powerful free radical scavengers PTIO and 2-MPG did not show any beneficial effect on SNP toxicity in our experimental setting.
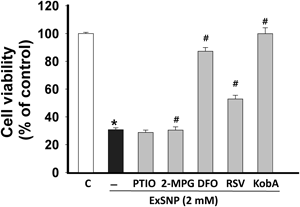
Apart from liberating NO from SNP, iron overloading may occur and this may lead a severe cell death.16) When the iron overloading was inhibited by DFO, the cellular toxicity of SNP was dramatically decreased (Fig. 5). To further test if iron overloading is crucial for SNP-induced H9c2 cell death, we assessed the toxicity of exhausted SNP (ExSNP) produced by photodegradation of SNP.21) NO production from ExSNP was determined using a NO-specific fluorescent dye (Fig. 6). When compared to SNP, ExSNP showed a significant decrease in the release of NO when compared to SNP (SNP vs. ExSNP; 260.5±9.9 vs. 25.7±1.2%, p<0.05) although it releases a small amount of NO. Despite the attenuated release of NO from ExSNP, the cytotoxicy of ExSNP was similar to that of SNP (SNP vs. ExSNP; 26.4±0.8 vs. 30.8±1.4%, p<0.05). PTIO, RSV and Kob A also showed protection similar to that of SNP (Fig. 7). These results suggest that the main cause of SNP-induced cell death may rely on the excessive iron moiety released from SNP or ExSNP (Figs. 6, 7). In this study, resveratrol showed the protective effect against SNP- and ExSNP-induced toxicity which was produced by photodegradation and released less NO, implying that RSV may also have a capacity of iron modulation in cells (Figs. 5, 7). However, further study is required to confirm this activity of RSV.

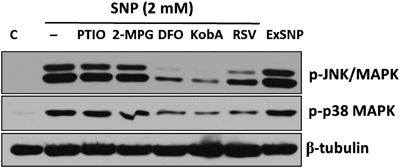
It has been known that SNP exposure activates cellular stress responses and this leads to the activation of p38 mitogen-activated protein kinase (MAPK) and JNK in the H9c2 cells.5,22,23) Among the activated MAPKs exposed to SNP, JNK was strongly involved in cardiac cell death, since the dominant negative form of JNK dramatically suppressed SNP-induced cell death.5,23) Thus we evaulated the changes in the phosphorylation levels of JNK and p38 MAPK following SNP or ExSNP treatments in the presence or absence of PTIO, 2-MPG, DFO, RSV, and Kob A. As shown in Fig. 8, SNP and ExSNP greatly increased phosphorylation of both JNK and p38 MAPK. In accordance with the cell viability results (Figs. 5, 7), both PTIO and 2-MPG did not attenuate the activation of JNK and p38 MAPK. However, DFO, RSV, and Kob A which showed the protective activity against excessive SNP-induced cytotoxicity suppressed the phosphorylation of both JNK and p38 MAPK. Based on these results, the toxic effect of high SNP may stem from a released redox-active iron moiety within H9c2 cells and iron-related moiety may be, at least, involved in the phosphorylation of both JNK and p38 MAPK. However, the direct action of Kob A on modulating the behavior of iron from SNP needs to be investigated in the future experiments.
Caspase-3, which is a common protease in both mitochondria and death receptor-dependent pathways, is known as a final effector in apoptotic cell death.24) In various cell lines, SNP-induced cell death is related to cytochrome c release from mitochondria and caspase-3 activation, which is one of the key executioners of apoptosis.5,25,26) The activation of caspase-3 requires proteolytic processing of its inactive zymogen into activated p17 and p12 fragments. When the activation of caspase-3 was measured (Fig. 9), SNP robustly increased the density of the cleaved form of caspase-3; but this increase was powerfully inhibited in presence of Kob A (SNP; 198.1±8.3% vs. SNP with Kob A; 32.0 ±6.1%, p<0.05).
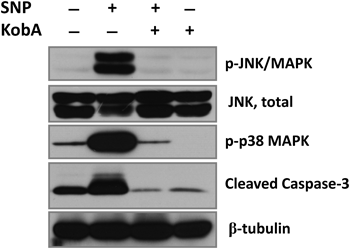
When the activation of JNK was measured by the level of its phosphorylated form, phosphorylation of JNK was significantly attenuated in the presence of Kob A (SNP; 598.6±68.5% vs. SNP with Kob A; 127.6±13.35%, p<0.05), but Kob A per se did not affect the phosphorylation of JNK (Fig. 9). This result implies that SNP-induced activation of JNK, which may lead to H9c2 cell death, was blocked by Kob A (Figs. 8, 9). Exposure to SNP also dramatically activated p38 MAPK which resulted in an increase of phosphorylation of p38 MAPK in cardiac H9c2 cells (SNP; 437.7±10.7% vs. SNP with Kob A; 84.1±0.1%, p<0.05). The p38 MAPK not only plays a role in sensing cellular stresses and participates in the control of cell death, but also can be involved in the cell cycle regulation.13,14) Contrary to a previous finding showing the activation of p38 MAPK in human osteoblast-like cells,27) in present study Kob A suppressed SNP-mediated phosphorylation of p38 MAPK in H9c2 cells. In addition, Kob A alone did not increase the phosphorylation of p38 MAPK (Fig. 9). Although the exact reason is unclear, it can be postulated that the different effects of Kob A on p38 MAPK may be dependent on the cellular status and/or conditions.
JNK can regulate members of the Bcl-2 protein family, which are important factors in mitochondria-mediated cell death, making mitochondria another target of pro-apoptotic signaling by JNK.28,29) The down regulation of Bcl-2 and Mcl-1 by oxidative stress insults is an indirect indicator of mitochondrial-mediated damages, since these two anti-apoptotic proteins play a role in protecting mitochondria against toxic agents. Next, we tested whether Kob A could prevent SNP-induced reduction of Bcl-2 and Mcl-1 levels, which are important mitochondrial anti-apoptotic proteins. As expected, SNP treated H9c2 cells showed a significant decrease in cellular levels of Bcl-2 (Fig. 10). In accordance with the decrease in SNP-induced phosphorylation of JNK by Kob A, the down-regulation of Bcl-2 was inhibited in the presence of Kob A (SNP; 59.8±4.0% vs. SNP with Kob A; 98.8±4.3%, p<0.05). Mcl-1 is unique among other members of the pro-survival Bcl-2 molecules due to its short protein half-life and promotion of the survival of a myriad of cellular lineages. Thus, the loss or reduction of Mcl-1 protein can lead to cell death.30) After exposure of cells to stress, Mcl-1 can be rapidly degraded via a proteasomal pathway and this destruction can be regulated by JNK and glycogen synthase kinase 3.31) Treatment of SNP severely reduced the protein level of Mcl-1, and this also may be related to SNP-induced cell death. Kob A could sufficiently preserve Mcl-1 protein in SNP-exposed cells (SNP; 48.7±7.1% vs. SNP with Kob A; 78.43±7.2%, p<0.05). However, it is unclear whether this protective effect of Kob A is tightly related to suppression of JNK.

Taken together, a high dose of SNP severely damaged cardiac H9c2 cells but Kob A provided a protective activity against SNP-induced toxicity. This beneficial effect of Kob A may be, at least in part, related to the blocking of activation of JNK and p38 MAPK, and preservation of mitochondrial anti-apoptotic proteins in H9c2 cells. This property of Kob A may provide an insight into developing new cardioprotectant against SNP-mediated cardiac cell death.
Experimental
General Experimental ProceduresMelting point was measured on a Gallenkamp melting point apparatus (uncorr.). 1H- and 13C-NMR were performed on a Varian Unity INOVA 500 spectrometer with the usual pulse sequences. NMR signals are reported in parts per million (δ), referenced to acetone-d6. HPLC was performed using a Phenomenex® Gemini 5µC18 110 A (150×4.60 mm) column and a UV detector (254 nm).
Plant MaterialThe roots of Caragana sinica (C. sinica) were collected at Changnyeong-gun, Gyeongsangnam-do, Korea, in May 2005, and identified by Prof. Kwak J. H., Sungkyunkwan University, Republic of Korea. The voucher specimen (SKKU-PH-05-83) is deposited in the School of Pharmacy, Sungkyunkwan University, Republic of Korea.
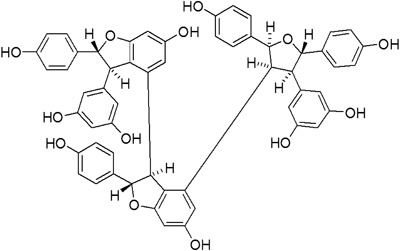
Extraction and Isolation of Kob AThe dried roots of C. sinica (500 g) were extracted three times with MeOH at room temperature. The MeOH extract was evaporated to dryness under reduced pressure, and the residue (37 g) was suspended in H2O. The resulting solution was partitioned consecutively with CH2Cl2, EtOAc, and n-BuOH. The EtOAc extract was fractionated on a Lipophilic Sephadex LH-20 (Sigma Chemical Co., St. Louis, MO, U.S.A.) column with MeOH. The fractions were combined based on their TLC pattern to yield fractions F1–F7. Fraction F3 (2.45 g) was rechromatographed over a MCI gel CHP 20P (75–150 µm, Mitsubishi Chemical Co., Tokyo, Japan) column with 80% MeOH, and then further rechromatographed on a Lichroprep® RP-18 (40–63 µm, MERCK, Darmstadt, Germany) column using 43% MeOH as the eluent to obtain Kob A (1.05 g). Kob A was identified by comparing its NMR spectral data with the literature values.9) The purity of the isolated compound was confirmed to be over 98.3% by HPLC analysis, and the chemical structure of Kob A is shown in Fig. 1. The chemical structure of Kob A is [3,4′-bibenzofuran]-6,6′-diol,3′-(3,5-dihydroxyphenyl)-4-[(2S,3S,4R,5S)-4-(3,5-dihdroxyphenyl)tetrahydro-2,5-bis(4-hydroxyphenyl)-3-furanyl]-2,2′,3,3′-terahydro-2,2′-bis(4-hydroxyphenyl)-,(2S,2′R,3S,3′R). Kob A was dissolved in dimethyl sulfoxide as a 25 mM stock solution and diluted to the appropriate concentrations with the culture medium.
Kob A: Brownish amorphous solid; mp 232–234°C; 1H-NMR (Me2CO-d6, 500 MHz) δ: 3.10 (1H, dd, J=10.7, 6.2 Hz, H-8d), 3.29 (1H, dd, J=6.0, 4.2 Hz, H-8c), 3.49 (1H, d, J=3.3 Hz, H-8b), 4.31 (1H, d, J=1.8 Hz, H-8a), 5.00 (1H, d, J=3.6 Hz, H-7b), 5.04 (1H, d, J=4.2 Hz, H-7c), 5.15 (1H, d, J=10.2 Hz, H-7d), 5.51 (1H, br s, H-12b), 5.80 (2H, d, J=2.4 Hz, H-10d, 14d), 5.96 (1H, d, J=2.1 Hz, H-14b), 5.99 (1H, d, J=2.4 Hz, H-12c), 6.02 (3H, s, H-10a, 12a, 14a), 6.08 (1H, t, J=2.1 Hz, H-12d), 6.20 (2H, d, J=8.7 Hz, H-2b, 6b), 6.40 (2H, d, J=8.4 Hz, H-2c, 6c), 6.41 (1H, d, J=2.1 Hz, H-14c), 6.49 (2H, d, J=8.4 Hz, H-3b, 5b), 6.51 (1H, d, J=1.8 Hz, H-12b), 6.59 (2H, d, J=8.4 Hz, H-3c, 5c), 6.76 (2H, d, J=8.4 Hz, H-3d, 5d), 6.88 (2H, d, J=8.7 Hz, H-3a, 5a), 7.07 (2H, d, J=8.4 Hz, H-2d, 6d), 7.33 (2H, d, J=8.4 Hz, H-2a, 6a); 13C-NMR(Me2CO-d6, 125 MHz) δ: 52.2 (C-8b), 52.4 (C-8c), 58.1 (C-8a), 62.1 (C-8d), 84.8 (C-7c), 85.1 (C-7d), 92.4 (C-7a), 93.3 (C-7b), 95.7 (C-12c), 96.1 (C-12b), 102.0 (C-12a), 103.4 (C-12d), 106.6 (C-10a, 14a), 108.3 (C-14b), 109.1 (C-10d, 14d), 110.3 (C-14c), 115.3 (C-3c, 5c), 115.8 (C-3b, 5b), 115.9 (C-3d, 5d), 116.4 (C-3a, 5a), 119.9 (C-10b), 124.2 (C-10c), 126.6 (C-2a, 6a), 127.2 (C-2b, 6b), 127.5 (C-2c, 6c), 128.7 (C-2d, 6d), 131.8 (C-1c), 133.6 (C-1b), 134.4 (C-1d), 135.3 (C-1a), 136.2 (C-9c), 139.3 (C-9d), 144.6 (C-9b), 147.5 (C-9a), 156.0 (C-4c), 157.3 (C-4b), 157.7 (C-4d), 158.0 (C-4a), 158.2 (C-11d, 13d), 158.6 (C-13c), 159.5 (C-11a, 13a), 160.7 (C-13b), 160.9 (C-11c), 161.8 (C-11b).
Cell Culture and SNP TreatmentRat cardiac-like H9c2 cells (ATCC, Rockville, MD, U.S.A.) were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% heat-inactivated fetal bovine serum, L-glutamine (2 mM), penicillin (50 IU/mL), and streptomycin (50 µg/mL) (Gibco-BRL, Grand Island, NY, U.S.A.). Sodium nitroprusside (Sigma, St. Louis, MO, U.S.A.) was dissolved in D-PBS at a concentration of 1 mol and sterilized through a 0.2 µm filter before use. ExSNP was prepared by photodegradation under light at room temperature at least for 2 d.21)
Measurement of Cell ViabilityExponentially growing H9c2 cells were seeded at a cell density of 2×104 cells/well in 96-well tissue culture plates and treated with 2 mM SNP in absence or presence of various concentrations of Kob A. Cell viability was measured by quantitative colorimetric assay with MTT, showing the mitochondrial activity of living cells. The extent of the reduction of MTT to formazan within cells was quantified by measuring the optical density at 550 nm using a Molecular Device microplate reader (Menlo Park, CA, U.S.A.). The cell viability was expressed as a percentage of the untreated controls.
Assessment of NO ProductionThe amount of NO2− accumulated in the culture supernatants was measured using the assay system reported.11) Griess reagent was prepared by mixing one part 0.1% naphthylethylenediamine dihydrochloride in distilled water plus one part 1% sulfanilamide in 5% concentrated H3PO4. Briefly, 100 µL supernatant was removed from each well into an empty 96-well plate. After the addition of 100 µL Griess reagent to each well, absorbance at 540 nm was measured using a Molecular Device microplate reader. Nitrite concentration was calculated from a NaNO2 standard curve. For measuring the changes in NO production from SNP32), H9c2 cells were cultured until reached to confluency in a 48 well tissue culture plates (Nunc, Denmark). After washing twice with Tyrode solution containing (in mM) NaCl 135, KCl 5.4, N-(2-hydroxyethyl)piperazine-N′-2-ethanesulfonic acid (HEPES) 5, NaH2PO4 0.33, MgCl2 0.5, glucose 16.6, and CaCl2 1.8, pH 7.4, cells were loaded with 4-amino-5-methylamino-2′,7′-difluorofluorescein (DAF-FM) (10 µM) for 20 min at 37°C. After washing three times with Tyrode solution, cells were filled with fresh Tyrode solution. Then SNP or ExSNP (2 mM) were loaded onto cells. The intensity of DAF-FM fluorescence (excitation/emission; 495/515 nm)32) was measured using SpectraMax M2e (Molecular Devices, OR, U.S.A.) with scan mode before and 30 min after adding the SNP or ExSNP. The DAF fluorescent change was calculated as the % increase of baseline. SNP-untreated group was used as a negative control.
Western Blot AnalysisH9c2 cells were incubated with 10 mL medium containing 2 mM SNP in the presence or absence of Kob A (25 µM) at 37°C for various times in 100 mm culture dishes. The cells were washed twice with cold D-PBS and then scraped in the presence of radio immunoprecipitation assay (RIPA) buffer (25 mM Tris–HCl pH 7.6, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS)) including protease/phosphatase inhibitor cocktails (Sigma). Equal amounts of protein (50 µg) were electrophoresed on 10% or 12% SDS-polyacrylamide gel and transferred to an Immobilon®-P polyvinylidene difluoride membrane. The binding of the primary antibody was detected with a secondary antibody and then visualized using the enhanced chemiluminescence (ECL) method. Equal loading of samples was confirmed by re-probing the membranes with anti-β-tubulin antibody. The band density from Western blotting was measured using Multi Gauge Ver. 3.0 software (FUJIFILM, Japan).
Statistical AnalysisStatistical calculations were performed with SigmaStat (Systat software) for Windows software package. Each result is reported as means±S.E.M. All experiments were repeated at least three times independently and the significant values are set at p<0.05 with one-way ANOVA test.
Supporting Information1H- and 13C-NMR spectra of kobophenol A are available as Supporting Information.