Because of diverse and prominent biological activities, quinazoline derivatives still draw attention from synthetic and pharmaceutical chemists.1–3) Among the compounds containing a quinazoline core, the most widely used are tyrosine kinase inhibitor anticancer drugs, such as Gefitinib (1),4) Erlotinib (2),5) and Lapatinib (3),6) currently used as targeted chemotherapy drugs for advanced or metastatic nonsmall cell lung and breast cancers (Fig. 1). Moreover, α-adrenergic receptor antagonist antihypertensive drugs such as Alfuzosin (4)7) and Prazosin (5)8) also contain a quinazoline core (Fig. 1). Quinazoline-2,4(1H,3H)-diones also exhibit various pharmacological functions such as antibacterial,9,10) anticonvulsant,9) anti-inflammatory,11,12) anticancer,13,14) and analgesic.12) Moreover, according to the most recent results, quinazoline-2,4(1H,3H)-diones also exhibit other significant pharmacological functions such as vascular endothelial growth factor receptor 2 (VEGFR-2) receptor tyrosine kinase inhibitor,15) heat shock protein 90 inhibitor,16) glucocerebrosidase inhibitor,17) and antitumor.18–20)
A substantial amount of reports on quinazoline-2,4(1H,3H)-diones have been published because of their diverse and vital biological applications.1,21) In summary, the existing synthetic methods can be grouped into four categories (Chart 1): (1) The carbon fixation reaction of anthranilonitriles with high-pressure CO2 catalyzed by strong organic bases or transition metals at high temperatures afforded quinazoline-2,4(1H,3H)-diones.22–28) Recently, Li et al. reported a high-temperature ZnCl2-catalyzed coupling/cyclization of anthranilonitrile with DMF to form quinazoline-2,4(1H,3H)-diones.29) (2) The reactions of anthranilic acid and its esters with carbamate afforded the corresponding urea derivatives; subsequent cyclization reactions afforded quinazoline-2,4(1H,3H)-diones.20,30) (3) Anthranilamide is also an important starting material for the preparation of quinazoline-2,4(1H,3H)-diones via urea. The desired compounds were obtained by direct carbonylation/cyclization of urea.31,32) (4) Isatoic anhydride has also been successfully transformed to quinazoline-2,4(1H,3H)-diones.33) Although numerous synthetic methods for quinazoline-2,4(1H,3H)-diones have been reported, they suffer from several disadvantages such as high temperature,13,29,34,35) high pressure,22,27) transition-metal catalysis,36–38) and expensive reagents.26,37) During our research on anticancer drugs,39,40) a rapid and easy access to quinazolinediones was needed. Herein, we report an eco-efficient, environment-friendly, and one-pot method for the synthesis of quinazoline-2,4(1H,3H)-diones.

Chart 1. Representative Synthetic Methods for Quinazoline-2,4(1H,3H)-diones
Results and Discussion
To optimize the reaction conditions for the synthesis of quinazoline-2,4(1H,3H)-diones, 4-fluoro-2-aminobenzoic acid (1a) was used as the model compound, and the yields were determined by LC-MS analyses (Chart 2, Table 1). There are two crucial steps in the conversion from 1a to 2a. In the first step, the coupling of 1a with potassium cyanate (KOCN) affords the corresponding urea derivative (3a). In the second step, the cyclization of 3a in the presence of a base affords the cyclized intermediate, 4a. First, acetonitrile was used as the solvent because of the good solubility of the reactants in this solvent (Table 1, entries 1–5). Acetic acid (2.0 eq) was added to transform potassium cyanate (KOCN) into cyanic acid (HOCN). After 2 h at 50°C, the LC-MS analysis showed the formation of 85% 4-fluoro-2-ureidobenzoic acid (3a) in the reaction mixture, and 5% of 1a still existed (Table 1, entry 1). After decreasing the reaction temperature from 50°C to room temperature (25°C) and increasing the reaction time to 4 and 6 h, intermediate 3a could be obtained in 88% and 94% yields, respectively, with no starting material remaining after 6 h (Table 1, entries 2, 3). Then, NaOH was added directly into the reaction mixture; a complete cyclization of 3a to 4a (monitored by LC-MS) was achieved after the addition of 4 equiv NaOH. The pH of the reaction mixture was adjusted to pH 1 using concentrated HCl. After the evaporation of solvent and column purification, 7-fluoroquinazoline-2,4(1H,3H)-dione (2a) was obtained in 91% yield (Table 1, entry 5).

Chart 2. Synthesis of 7-Fluoroquinazoline-2,4(1H,3H)-dione (2a)
Table 1. Optimization of Reaction Conditions
a)| Entry | Solvent | T (°C) | Time | NaOH | Yield |
|---|
| 1 | CH3CN | 50 | 2 h | — | 85%b) |
| 2 | CH3CN | 25 | 4 h | — | 88%b) |
| 3 | CH3CN | 25 | 6 h | — | 94%b) |
| 4 | CH3CN | 25 | 6 h | 2.0 | 56%c) |
| 5 | CH3CN | 25 | 6 h | 4.0 | 91%c) |
| 6 | H2O | 85 | 6 h | — | 94%b) |
| 7 | H2O | 50 | 6 h | — | 95%b) |
| 8 | H2O | 25 | 6 h | — | 56%b) |
| 9 | H2O | 25 | 12 h | — | 92%b) |
| 10 | H2O | 25 | 12 h | 4.0 | 90%c) |
a) 4-Fluoro-2-aminobenzoic acid (5 g, 32.2 mmol, 1.0 eq) and AcOH (3.0 mL, 53.4 mmol, 1.8 eq) were dissolved in solvent (50 mL), the solution of potassium isocyanate (3.9 g, 44.5 mmol, 1.5 eq) was dropping slowling. After complete consumption starting material, NaOH was added in portions to cyclize the intermediate 3a. Acidification and filtration of the reaction mixture afforded desired product. b) Yields of the intermediate 3a. c) Yields of 7-fluoroquinazoline-2,4(1H,3H)-dione (2a).
After developing a reliable method to synthesize 2a in a high yield using acetonitrile as the solvent, we investigated the possibility of using a more eco-friendly solvent, water, instead of acetonitrile. The solubility of 1a in water is poor at room temperature. However, 5.0 g of 1a could be dissolved in 100 mL of water at 80°C. After the reaction mixture was acidified with 2 eq AcOH, an aqueous solution of KNCO (1.5 eq) was added dropwise. The reaction was completed after stirring at 80°C for 6 h (Table 1, entry 6). When the reaction was performed at room temperature in water, the reaction mixture formed a suspension initially. To our surprise, the LC-MS analysis showed the formation of urea after adding KNCO. The transformation was completed after 12 h affording 3a in a 92% yield (Table 1, entry 9), which is comparable with that using acetonitrile as the solvent (91% yield, Table 1, entry 5). After a slow addition of 4 eq NaOH, following which the pH was adjusted to below 1, compound 2a was obtained as a white solid after filtration. The purity of 2a was >98% according to the LC-MS analysis. Using this method, we were able to scale up 2a to 1 kg of starting material without any problem in 82% yield. In short, 2a was successfully synthesized at room temperature using water as the solvent. Moreover, the entire procedure is very convenient and efficient, and the desired product could be afforded in high yield and purity without additional purification.
After the successful synthesis of 2a, we investigated the substrate scopes of this method. o-Aminobenzoic acid with different electron-withdrawing or -donating substituents were used as the starting materials for the reaction with KOCN at room temperature in water, following which cyclization was performed in the presence of base and the product was filtered after acidification. Different quinazoline-2,4(1H,3H)-dione derivatives were obtained in high-to-excellent yields (Table 2). Products with electron-donating substituents on the benzene ring were obtained in a slightly higher yield (e.g., 88% yield, Table 2, entry 3). In contrast, products with electron-withdrawing substituents on the benzene ring were obtained in a slightly lower yield (e.g., 75% yield, Table 2, entry 9). Moreover, the position of the substituents also affected the reaction. The presence of ortho/para-electron-donating OMe substituents increased the yield of the product (95% yield, Table 2, entry 4) compared to that with an ortho-electron-withdrawing Cl substituent (82% yield, Table 2, entry 5). Notably, the reaction of 2-aminonicotinic acid afforded pyrido[2,3-d]pyrimidine-2,4(1H,3H)-dione (Table 2, entry 10) in 82% yield under these reaction conditions, thus providing a new synthetic method for the preparation of this novel heterocyclic compound.
Next, further synthetic transformations of the quinazolinediones were investigated. The chlorination of 2a with phosphorus oxychloride afforded a doubly chlorinated product, 2,4-dichloro-7-fluoroquinazoline (6). The structure of 6 was confirmed by single-crystal X-ray diffraction analysis (Fig. 2).41) The selective reduction of the Cl group at the C-4 position on 6 with Zn powder afforded 2-chloro-7-fluoroquinazoline (7) in a high yield, thus paving the way for the synthesis of active quinazoline compounds with different substituents at the C-2 position on the quinazoline ring (Chart 3). Moreover, the selective hydrogenation/reduction of the Cl group at the C-4 position with Zn powder was also investigated.

Chart 3. Synthesis of 2-Chloro-7-fluoroquinazoline and Its Possible Applications
Experimental
General MethodNMR spectra were recorded in DMSO-d6 using a Varian INOVA-400/54 high-resolution superconducting NMR; Tetramethylsilane (TMS) was used as the internal standard. The crystal structure was determined using a four-circle X-ray diffractometer. An Agilent 1260–6120 LCMS was used to obtain the LC-MS data.
Typical Experimental ProcedureAn aqueous solution of KOCN (75 mmol dissolved in 20 mL water) was added dropwise into a mixture of anthranilic acid derivative (50 mmol) and AcOH (90 mmol) in deionized water (80 mL). The reaction mixture was stirred overnight, and then solid NaOH (200 mmol) was added in portions. After stirring for another 4 h, the pH of the reaction mixture was carefully adjusted to pH <1 with concentrated HCl, affording a white precipitate. After filtration and washing with water until pH 7 was attained, the desired products were obtained. The structures of the products were confirmed by spectral analyses, and the data agreed well with those reported in the literature.
Characterization Data of Products7-Fluoroquinazoline-2,4(1H,3H)-dione (2a): White solid, 1H-NMR (400 MHz, DMSO-d6) δH: 11.34, 11.24 (each 1H, s), 7.95 (1H, dd, J1=11.2 Hz, J2=8.8 Hz), 7.01 (1H, m), 6.88 (1H, dd, J1=8.8 Hz, J2=2.8 Hz) ppm; 13C-NMR (100 MHz, DMSO-d6) δC: 165.7 (d, J=332 Hz), 161.9, 150.2, 142.8 (d, J=16 Hz), 130.1 (d, J=16 Hz), 113.2 (d, J=2 Hz), 110.2 (d, J=32 Hz), 101.4 (d, J=35 Hz) ppm. High resolution-electrospray ionization (HR-ESI)-MS: Calcd for C8H6FN2O2, 181.0413, Found 181.0418 [M+H]+.
Quinazoline-2,4(1H,3H)-dione (2b): White solid, 1H-NMR (400 MHz, DMSO-d6) δH: 11.31, 11.17 (each 1H, s), 7.91 (1H, dd, J1=8.4 Hz, J2=1.2 Hz, H-Ar), 7.66 (1H, m), 7.20 (2H, m); 13C-NMR (100 MHz, DMSO-d6) δC: 162.9, 150.3, 140.9, 134.9, 126.9, 122.4, 115.3, 114.4 ppm. The NMR data for this compound matched the literature data.23,25,27)
7-Methylquinazoline-2,4(1H,3H)-dione (2c): White solid, 1H-NMR (400 MHz, DMSO-d6) δH: 11.20, 11.04 (each 1H, s), 7.69 (s), 7.45 (1H, dd, J1=8.0 Hz, J2=1.2 Hz), 7.07 (1H, d, J=8.0 Hz) 2.36 (s, 3H) ppm; 13C-NMR (100 MHz, DMSO-d6) δC: 162.8, 150.3, 145.6, 140.7, 126.8, 123.5, 115.0, 112.1, 21.3 ppm. The NMR data for this compound matched the literature data.29)
5,6,7-Trimethoxyquinazoline-2,4(1H,3H)-dione (2d): White solid, 1H-NMR (400 MHz, DMSO-d6) δH: 11.10, 11.08 (each 1H), 6.52 (s); 3.84 (3H, s), 3.78 (3H, s), 3.70 (3H, s) ppm; 13C-NMR (100 MHz, DMSO-d6) δC: 160.8, 158.7, 154.3, 150.5, 139.9, 138.1, 102.2, 94.4, 62.2, 61.3, 56.4 ppm. The NMR data for this compound matched the literature data.42)
8-Chloroquinazoline-2,4(1H,3H)-dione (2e): Yellow solid, 1H-NMR (400 MHz, DMSO-d6) δH: 11.65, 11.50 (each 1H, s), 7.87 (d, J=8.0 Hz), 7.77 (1H, d, J=8.0 Hz), 7.17 (1H, t, J=8.0 Hz); 13C-NMR (100 MHz, DMSO-d6) δC: 161.9, 151.4, 137.8, 133.5, 130.6, 126.6, 125.4, 115.4 ppm. The NMR data for this compound matched the literature data.20)
7-Chloroquinazoline-2,4(1H,3H)-dione (2f): Yellow solid, 1H-NMR (400 MHz, DMSO-d6) δH: 11.45, 11.24 (each 1H, s), 7.89 (d, J=8.4 Hz), 7.25 (1H, d, J=8.4 Hz), 7.18 (1H, s) ppm; 13C-NMR (100 MHz, DMSO-d6) δC: 162.2, 150.1, 141.9, 139.4, 128.9, 122.4, 114.6, 113.3 ppm. The NMR data for this compound matched the literature data.25,27)
6-Bromoquinazoline-2,4(1H,3H)-dione (2g): Yellow solid, 1H-NMR (400 MHz, DMSO-d6) δH: 11.46, 11.28 (each 1H), 7.94 (s, 1H), 7.78 (1H, d, J=8.4 Hz), 7.11 (1H, d, J=8.4 Hz) ppm; 13C-NMR (100 MHz, DMSO-d6) δC: 161.7, 150.0, 140.0, 137.4, 128.9, 117.7, 116.2, 113.7 ppm. The NMR data for this compound matched the literature data.25,27,29)
6-Nitroquinazoline-2,4(1H,3H)-dione (2i): White solid, 1H-NMR (400 MHz, DMSO-d6) δH: 11.45, 11.32 (each 1H, s), 7.93 (s, 1H), 7.80 (1H, dd, J1=8.4 Hz, J2=1.6 Hz), 7.14 (1H, d, J=8.4 Hz); 13C-NMR (100 MHz, DMSO-d6) δC: 161.9, 150.2, 140.3, 137.6, 129.0, 118.0, 116.3, 114.0 ppm. The NMR data for this compound matched the literature data.43)
Pyrido[2,3-d]pyrimidine-2,4(1H,3H)-dione (2j): Yellow solid, 1H-NMR (400 MHz, DMSO-d6) δH: 11.66, 11.46 (each 1H, s), 8.16 (d, 1H, J=4.4 Hz), 8.02 (1H, d, J=7.7 Hz), 6.60 (1H, dd, J1=4.4 Hz, J2=7.6 Hz); 13C-NMR (100 MHz, DMSO-d6) δC: 161.5, 152.9, 150.4, 146.3, 139.7, 122.9, 115.3 ppm. The NMR data for this compound matched the literature data.43)
2,4-Dichloro-7-fluoroquinazoline (6): Yellow crystal from ethyl acetate, mp 145–147°C, 1H-NMR (400 MHz, DMSO-d6) δH: 8.42 (1H, m), 7.95 (1H, d, J=3.2 Hz), 7.92 (1H, m); The yellow sheet crystal of 6 from acetone was mounted on a P4 four-circle diffractometer and exposed to graphite-monochromated Mo Kα irradiation. The unit cell parameters are a=3.8257 (3) Å, b=15.0664 (9) Å, c=14.3453 (6) Å in space group P21/n (Z=4), Dx=1.750 g/cm3. The structure was solved by direct methods with the program SHELX 97 and refined by full-matrix least-squares on F2. The final R indices were R1=0.038 and wR2=0.095. CCDC979379 contains the supplementary crystallographic data for this compound. These data can be obtained free of charge via https://www.ccdc.cam.ac.uk/.
2-Chloro-7-fluoroquinazoline (7): Yellow solid, 1H-NMR (400 MHz, DMSO-d6) δH: 9.20 (1H, s), 7.93 (1H, dd, J1=11.6 Hz, J2=8.0 Hz), 7.54 (1H, dd, J1=8.0 Hz, J2=2.8 Hz), 7.39 (1H, ddd, J1=11.6 Hz, J2=8.0 Hz, J3=2.8 Hz) ppm. The 1H-NMR data for this compound matched the literature data.44)


 1a
1a 2a
2a 1b
1b 2b
2b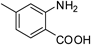 1c
1c 2c
2c 1d
1d 2d
2d 1e
1e 2e
2e 1f
1f 2f
2f 1g
1g 2g
2g 1h
1h 2h
2h 1i
1i 2i
2i 1j
1j 2j
2j