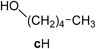Experimental
Melting points were determined using a micro melting point apparatus (Yanaco MP-S3) without correction. IR spectra were measured by a Shimadzu FTIR-8100 IR spectrophotometer. Low- and high-resolution mass spectra (LR-MS and HR-MS) were obtained by a JEOL JMS HX-110 double-focusing model equipped with an FAB ion source interfaced with a JEOL JMA-DA 7000 data system. lH- and 13C-NMR spectra were obtained by JEOL JNM A-500. Chemical shifts were expressed in δ ppm downfield from an internal TMS signal for lH-NMR and the carbon signal of the corresponding solvent [chloroform-d3 (CDCl3) (77.00 ppm), methanol-d4 (CD3OD) (49.00 ppm)] for 13C-NMR. The abbreviations qu=quintet, dd=double doublets, and dm=double multiplets are used for the multiplicity of 1H-NMR data. The signal assignments were confirmed by two-dimensional (2D)-NMR analyses: 1H–1H 2D correlation spectroscopy (COSY), lH–l3C heteronuclear multiple-quantum coherence (HMQC), and 1H–l3C heteronuclear multiple-bond connectivity (HMBC). Microanalyses were performed with a Yanaco MT-6 CHN corder. Routine monitoring of reactions was carried out using precoated Kieselgel 60F254 plates (E. Merck). Open and flash column chromatography separations of the reaction products were performed on silica gel (Kanto 60N) with a UV detector. Commercially available starting materials and compound 4a were used without further purification, and dry solvents were used in all reactions.
General Procedure for the Preparation of Trialkoxy-1,3,5-triazine Derivatives (Method A18)): Example: Preparation of 2,4,6-Tris(1-heptyloxy)-1,3,5-triazine (4d) (Entry 3): (Step 1)To a solution of n-heptanol (dH, 5.23 g, 45.0 mmol) in dry benzene (10 mL) was added NaH (60% in mineral oil, 1.26 g, 31.5 mmol) at room temperature under an N2 atmosphere. A suspension of sodium n-heptyloxide was prepared by stirring at room temperature for 0.5 h and then refluxing for 19 h. (Step 2) After cooling to room temperature, compound 1 (1.84 g, 10.0 mmol) in dry benzene (20 mL) was added dropwise with stirring. After stirring for 20 min at room temperature, the reaction mixture was refluxed for 1 h. After cooling to room temperature, the resulting mixture was acidified with acetic acid (ca. 2 mL) and then diluted with water (ca. 20 mL). The separated organic layer was washed with water (ca. 20 mL) and dried over magnesium sulfate (MgSO4). Evaporation of the solvent gave a pale yellow residual oil. To this residue was added n-hexane (ca. 10 mL), and insoluble solids were removed. The filtrate was purified by flash chromatography (n-hexane : EtOAc=97 : 3→95 : 5) to give 4d (2.91 g, 69%) as a colorless oil.
4d: IR (NaCl) cm−1: 1566 (C=N) 1145, 1118 (C–O of ether). 1H-NMR (CDCl3) δ: 0.88 (9H, t, J=7.0 Hz, H7′), 1.26–1.37 (18H, m, H4′, 5′, 6′), 1.38–1.46 (6H, m, H3′), 1.75–1.81 (6H, m, H2′), 4.38 (6H, t, J=6.7 Hz, H1′). 13C-NMR (CDCl3) δ: 13.98 (C7′), 22.53 (C6′), 25.76 (C3′), 28.64 (C2′), 28.91 (C4′), 31.69 (C5′), 68.45(C1′), 173.16 (C=N). Positive-ion FAB-MS m/z: 424 (M+H+). HR-FAB-MS m/z: 424.3547 (Calcd for C24H46N3O3: 424.3539). Anal. Calcd for C24H45N3O3: C, 68.04; H, 10.71; N, 9.92. Found: C, 67.88; H, 10.88; N, 9.91.
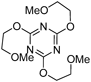
2,4,6-Tris(2-methoxyethoxy)-1,3,5-triazine (4b) (Entry 1)This compound was prepared from the reaction of compound 1 with 2-methoxyethanol (bH) by using Method A under the conditions shown in Table 1. Separation of the products by flash chromatography (n-hexane : EtOAc=35 : 65→30 : 70) gave 4b (41%) as a pale yellow oil.
4b: IR (NaCl) cm−1: 1569 (C=N), 1151, 1122 (C–O of ether). 1H-NMR (CDCl3) δ: 3.40 (9H, s, OCH3), 3.72 (6H, t, J=4.9 Hz, H2′), 4.54 (6H, t, J=4.9 Hz, H1′). 13C-NMR (CDCl3) δ: 58.91 (OCH3), 67.23 (C1′), 70.04 (C2′), 172.97 (C=N). Positive-ion FAB-MS m/z: 304 (M+H+). HR-FAB-MS m/z: 304.1498 (Calcd for C12H22N3O6: 304.1509). Anal. Calcd for C12H21N3O6·0.2H2O: C, 49.96; H, 7.03; N, 13.69. Found: C, 49.95; H, 6.93; N, 13.72.
2,4,6-Tris(1-pentyloxy)-1,3,5-triazine (4c)18) (Entry 2)This compound was prepared from compound 1 and 1-pentanol (cH) (Method A) under the conditions shown in Table 1. Separation of the reaction products by flash chromatography (n-hexane : EtOAc=93 : 7→95 : 5) gave 4c (74%) as a colorless oil.
4c: IR (NaCl) cm−1: 1567 (C=N), 1144, 1116 (C–O of ether). 1H-NMR (CDCl3) δ: 0.91 (9H, t, J=7.0 Hz, H5′), 1.32–1.45 (12H, m, H3′, 4′), 1.75–1.82 (6H, m, H2′), 4.38 (6H, t, J=6.7 Hz, H1′). 13C-NMR (CDCl3) δ: 13.85 (C5′), 22.30 (C4′), 27.91 (C3′), 28.30 (C2′), 68.40 (C1′), 173.14 (C=N). Positive-ion FAB-MS m/z: 340 (M+H+). HR-FAB-MS m/z: 340.2601 (Calcd for C18H34N3O3: 340.2600). Anal. Calcd for C18H33N3O3·0.4H2O: C, 62.36; H, 9.83; N, 12.12. Found: C, 62.47; H, 9.87; N, 12.34.
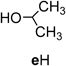
Preparation of 2,4,6-Tris(2-propoxy)-1,3,5-triazine (4e)8,18) (Entry 4)This compound was obtained in 77% yield from the reaction of compound 1 with isopropanol (eH) by using Method A under the conditions shown in Table 1. Recrystallization from EtOH gave analytically pure 4e as colorless crystals, mp 106–108°C (from EtOH). Spectral data (IR, NMR, and MS) and elemental analysis data of the product 4e were consistent with those of the authentic sample.8)
2,4,6-Tris(3-pentyloxy)-1,3,5-triazine (4f)18) (Entry 5)This compound was prepared from compound 1 and 3-pentanol (fH) (Method A) under the conditions shown in Table 1. Separation of the reaction products by flash chromatography (n-hexane : EtOAc=98 : 2→95 : 5) afforded 4f (72%) as colorless solids. An analytical sample of 4f was obtained by recrystallization from water as colorless crystals.
4f: mp 66–67°C (from H2O). IR (KBr) cm−1: 1553 (C=N), 1137, 1100 (C–O of ether). 1H-NMR (CD3OD) δ: 0.95 (18H, t, J=7.5 Hz, CH3), 1.69–1.76 (12H, m, CH2), 5.08 (3H, qu, J=6.1 Hz, CH). 13C-NMR (CD3OD) δ: 9.84 (CH3), 27.33 (CH2), 82.30 (CH), 174.60 (C=N). Positive-ion FAB-MS m/z: 340 (M+H+). HR-FAB-MS m/z: 340.2602 (Calcd for C18H34N3O3: 340.2600). Anal. Calcd for C18H33N3O3: C, 63.68; H, 9.80; N, 12.38. Found: C, 63.53; H, 9.84; N, 12.35.
Reaction of Compound 1 with 3-Pentanol (fH): Formation of N2,N2,N4,N4-Tetraethyl-6-(pentan-3-yloxy)-1,3,5-triazine-2,4-diamine (7), N2,N2,N4,N4,N6,N6-Hexaethyl-1,3,5-triazine-2,4,6-triamine (8),20) and N2,N2,N4,N4-Tetraethyl-6-methoxy-1,3,5-triazine-2,4-diamine (9)29) (Method B) (Entry 6)To a solution of compound 1 (1.84 g, 10.0 mmol) in dry tetrahydrofuran (THF) (20 mL) was added 3-pentanol (fH, 4.41 g, 50.0 mmol) and TEA (3.54 g, 35.0 mmol) at room temperature in an atmosphere of argon. After stirring for 19 h at room temperature, the resulting precipitates (TEA·HCl salt) were removed by filtration. After evaporation of the solvent, the products were separated by flash chromatography (n-hexane : EtOAc=99 : 1→95 : 5) to give 8 (8%) as a colorless oil, 7 (30%) as a colorless oil, and 9 (2%) as a pale yellow oil.
7: IR (NaCl) cm−1: 1568 (C=N), 1225, 1096 (C–O of ether). 1H-NMR (CDCl3) δ: 0.93 (6H, t, J=7.5 Hz, H1″, 5″), 1.16 (12H, t, J=7.0 Hz, H2′), 1.60–1.76 (4H, m, H2″, 4″), 3.56 (8H, q, J=7.0 Hz, H1′), 4.97–5.02 (1H, m, H3″). 13C-NMR (CDCl3) δ: 10.03 (C1″, 5″), 13.35 (C2′), 26.72 (C2″, 4″), 41.24 (C1′), 78.23 (C3″), 165.62 (C2, 4), 170.90 (C6). Positive-ion FAB-MS m/z: 310 (M+H+). HR-FAB-MS m/z: 310.2577 (Calcd for C16H32N5O: 310.2607). Anal. Calcd for C16H31N5O·0.5H2O: C, 60.34; H, 10.13; N, 21.99. Found: C, 60.37; H, 10.11; N, 22.10.
8: 1H-NMR (CDCl3) δ: 1.14 (18H, t, J=7.0 Hz, H2′), 3.53 (12H, q, J=7.0 Hz, H1′). 13C-NMR (CDCl3) δ: 13.47 (C2′), 40.98 (C1′), 164.75 (C=N). Positive-ion FAB-MS m/z: 295 (M+H+). HR-FAB-MS m/z: 295.2611 (Calcd for C15H31N6: 295.2610).
9: 1H-NMR (CDCl3) δ: 1.16 (12H, t, J=7.0 Hz, H2′), 3.56 (8H, qu, J=7.0 Hz, H1′), 3.87 (3H, s, OCH3). 13C-NMR (CDCl3) δ: 13.26 (C2′), 41.28 (C1′), 53.27 (OCH3), 165.54 (C4, 6), 170.92 (C2). Positive-ion FAB-MS m/z: 254 (M+H+). HR-FAB-MS m/z: 254.1982 (Calcd for C12H24N5O: 254.1981).
2,4,6-Tris(cyclopentyloxy)-1,3,5-triazine (4g) (Entry 7)This compound was obtained from the reaction of compound 1 with cyclopentanol (gH) (Method A) under the conditions shown in Table 1. After work-up of the reaction mixture, crude orange solid material 4g was obtained. Recrystallization from EtOAc gave 4g (38%) as colorless crystals.
4g: mp 146–148°C (from EtOAc). IR (KBr) cm−1: 1561 (C=N), 1129 (C–O of ether). 1H-NMR (CDCl3) δ: 1.60 (6H, m, H3′, 4′), 1.78–1.90 (12H, m, H3′, 4′, 2′, 5′), 1.92–2.00 (6H, m, H2′, 5′), 5.46 (3H, qu, J=3.1 Hz, H1′). 13C-NMR (CDCl3) δ: 23.77 (C3′, 4′), 32.71 (C2′, 5′), 80.59 (C1′), 172.68 (C=N). Positive-ion FAB-MS m/z: 334 (M+H+). HR-FAB-MS m/z: 334.2130 (Calcd for C18H28N3O3: 334.2131). Anal. Calcd for C18H27N3O3: C, 64.84; H, 8.16; N, 12.60. Found: C, 64.71; H, 8.21; N, 12.54.
2,4,6-Tris(cyclohexyloxy)-1,3,5-triazine (4h) (Entry 8)This compound was obtained from compound 1 and cyclohexanol (hH) (Method A) under the conditions shown in Table 1. After work-up of the reaction mixture, crude pale yellow solid material 4h was obtained. Recrystallization from EtOH afforded 4h (35%) as colorless crystals.
4h: mp 230–233°C (from EtOH). IR (KBr) cm−1: 1561 (C=N), 1126, 1010 (C–O of ether). 1H-NMR (CDCl3) δ: 1.24–1.33 (3H, m, H4′), 1.35–1.44 (6H, m, H3′, 5′), 1.54–1.62 (9H, m, H4′, 2′, 6′), 1.76–1.83 (6H, m, H3′, 5′), 1.97–2.03 (6H, m, H2′, 6′), 5.03–5.10 (3H, m, H1′). 13C-NMR (CDCl3) δ: 23.79 (C3′, 5′), 25.39 (C4′), 31.58 (C2′, 6′), 76.14 (C1′), 172.71 (C=N). Positive-ion FAB-MS m/z: 376 (M+H+). HR-FAB-MS m/z: 376.2602 (Calcd for C21H34N3O3: 376.2600). Anal. Calcd for C21H33N3O3: C, 67.17; H, 8.86; N, 11.19. Found: C, 67.11; H, 8.87; N, 11.15.
2,4,6-Tris(cycloheptyloxy)-1,3,5-triazine (4i) (Entry 9)This compound was prepared from the reaction of compound 1 and cycloheptanol (iH) by using Method A under the conditions shown in Table 1. After work-up of the reaction mixture, crude yellow solid material 4i was obtained. Recrystallization from EtOH gave 4i (45%) as colorless crystals.
4i: mp 175–177°C (from EtOH). IR (KBr) cm−1: 1562 (C=N), 1129 (C–O of ether). 1H-NMR (CDCl3) δ: 1.44–1.51 (6H, m, H3′, 6′), 1.56–1.62 (12H, m, H4′, 5′), 1.69–1.76 (6H, m, H3′, 6′), 1.79–1.86 (6H, m, H2′, 7′), 2.01–2.07 (6H, m, H2′, 7′), 5.23 (3H, m, H1′). 13C-NMR (CDCl3) δ: 22.80 (C3′, 6′), 28.36 (C4′, 5′), 33.66 (C2′, 7′), 78.63 (C1′), 172.58 (C=N). Positive-ion FAB-MS m/z: 418 (M+H+). HR-FAB-MS m/z: 418.3075 (Calcd for C24H40N3O3: 418.3070). Anal. Calcd for C24H39N3O3: C, 69.03; H, 9.41; N, 10.06. Found: C, 69.08; H, 9.61; N, 10.07.
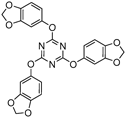
Procedure for the Synthesis of 2,4,6-(1,3-Benzodioxol-5-yloxy)-1,3,5-triazine (4j) (Method C) (Entry 10)To a solution of compound 1 (922 mg, 5.0 mmol) in dry acetone (25 mL) was added sesamol (jH, 2.14 g, 15.5 mmol) and collidine (1.88 g, 15.5 mmol) at room temperature. After stirring for 2 d at room temperature, the resulting precipitates were collected and the obtained material was added to CH2Cl2 (ca. 60 mL). The resulting mixture was stirred for 30 min at room temperature, and then the insoluble precipitates were collected and the obtained material was added to EtOH (ca. 150 mL). After stirring for 1 h at room temperature, the insoluble crude precipitates 4j were collected as a white solid (2.31 g, 95%). An analytical sample of 4j was obtained by recrystallization from acetone as colorless crystals.
4j: mp 249–250°C (from acetone). IR (KBr) cm−1: 1579 (C=N), 1175, 1140, 1040 (C–O). 1H-NMR (CDCl3) δ: 5.98 (6H, s, H2), 6.58 (3H, dd, J=8.2, 2.4 Hz, H6), 6.63 (3H, d, J=2.4 Hz, H4), 6.75 (3H, d, J=8.2 Hz, H7). 13C-NMR (CDCl3) δ: 101.80 (C2), 103.68 (C4), 107.92 (C7), 113.84 (C6), 145.63 (C7a), 146.00 (C5), 148.04 (C3a), 174.05 (C=N). Positive-ion FAB-MS m/z: 490 (M+H+). HR-FAB-MS m/z: 490.0888 (Calcd for C24H16N3O9: 490.0887). Anal. Calcd for C24H15N3O9: C, 58.90; H, 3.09; N, 8.59. Found: C, 58.92; H, 3.14; N, 8.61.
Procedure for the Synthesis of 2,4,6-[3-(Trifluoromethyl)phenoxy]-1,3,5-triazine (4k)30) (Method B) (Entry 11)A mixture of compound 1 (922 mg, 5.0 mmol), 3-(trifluoromethyl)phenol (kH, 2.51 g, 15.5 mmol), and TEA (1.57 g, 15.5 mmol) in dry acetone (20 mL) was stirred for 18 h at room temperature. After evaporation of the solvent, CH2Cl2 (ca. 100 mL) and 10% HCl (100 mL) were added to the reaction mixture. The separated organic layer was washed with brine (40 mL) and dried over MgSO4. Evaporation of the solvent gave crude white solid material 4k. Recrystallization from 2-PrOH gave analytically pure 4k (74%) as a colorless amorphous solid.
4k: mp 155–158°C (from 2-PrOH). IR (KBr) cm−1: 1572 (C=N), 1321 (C–F), 1183, 1124 (C–O). 1H-NMR (CDCl3) δ: 7.30 (3H, dm, J=7.6 Hz, H6′), 7.37 (3H, br s, H2′), 7.45–7.50 (6H, m, H4′, 5′). 13C-NMR (CDCl3) δ: 118.76 (q, J=4.1 Hz, C2′), 123.18 (q, J=4.1 Hz, C4′), 123.33 (q, J=272.1 Hz, CF3), 124.89 (C6′), 130.17 (C5′), 132.16 (q, J=33.1 Hz, C3′), 151.43 (C1′), 173.50 (C=N). Positive-ion FAB-MS m/z: 562 (M+H+). HR-FAB-MS m/z: 562.0823 (Calcd for C24H13F9N3O3: 562.0813). Anal. Calcd for C24H12F9N3O3: C, 51.35; H, 2.15; N, 7.49. Found: C, 51.11; H, 2.24; N, 7.57.
2,4,6-[4-(Trifluoromethyl)phenoxy]-1,3,5-triazine (4l) (Entry 12)This compound was prepared from the reaction of compound 1 and 4-(trifluoromethyl)phenol (lH) by using Method B under the conditions shown in Table 1. The resulting precipitates (TEA·HCl salt) were removed by filtration. After evaporation of the solvent, the products were separated by flash chromatography (n-hexane : CH2Cl2=4 : 6→1 : 9) to give 4l (33%) as a white solid. An analytical sample of 4l was obtained by recrystallization from 2-PrOH gave 4l as colorless needles.
4l: mp 217–218°C (from 2-PrOH). IR (KBr) cm−1: 1577 (C=N), 1331 (C–F), 1119, 1066 (C–O). 1H-NMR (CDCl3) δ: 7.23 (6H, d, J=8.5 Hz, H2′, 6′), 7.63 (6H, d, J=8.5 Hz, H3′, 5′). 13C-NMR (CDCl3) δ: 121.96 (C2′, 6′), 123.65 (q, J=272.0 Hz, CF3), 126.94 (q, J=4.1 Hz, C3′, 5′), 128.87 (q, J=33.1 Hz, C4′), 153.74 (C1′), 173.39 (C=N). Positive-ion FAB-MS m/z: 562 (M+H+). HR-FAB-MS m/z: 562.0807 (Calcd for C24H13F9N3O3: 562.0813). Anal. Calcd for C24H12 F9N3O3: C, 51.35; H, 2.15; N, 7.49. Found: C, 51.55; H, 2.15; N, 7.49.
General Procedure for the Preparation of Aryloxy-amino-1,3,5-triazine Derivatives (Method C): Example: Preparation of 1,1′-[6-[3-(Trifluoromethyl)phenoxy]-1,3,5-triazine-2,4-diyl]bis-4-piperidinemethanol (5kpp)(Step 1) A mixture of compound 1 (922 mg, 5.0 mmol), phenol (kH, 973 mg, 6.0 mmol), and collidine (727 mg, 6.0 mmol) in dry acetone (10 mL) was stirred for 10 min at 0°C and kept for 15 min at room temperature. The resulting colorless precipitated material (collidine·HCl) was removed by filtration and then the solvent was evaporated to afford a yellow solid. (Step 2) To this material in dry CH3CN (10 mL) was added 4-piperidinemethanol (pH, 2.30 g 20.0 mmol), and the resulting mixture was stirred for 1 h at room temperature. After evaporation of the solvent, a solvent (ca. 10 mL of n-hexane : 2-PrOH=80 : 20) was added to the residue, and then the insoluble materials were filtered to give crude 5kpp (1.92 g, 4.1 mmol, 81%) as a white solid. Recrystallization from propionitrile (EtCN) gave an analytically pure product 5kpp.
5kpp: mp 126–128°C (from EtCN). IR (KBr) cm−1: 1580 (C=N), 1323 (C–F), 1107, 1029 (C–O). 1H-NMR (CDCl3) δ: 1.10–1.25 (4H, m, H3′β, 5′β), 1.58 (2H, br s, OH), 1.68–1.82 (6H, m, H4′α, 3′α, 5′α), 2.72–2.85 (4H, m, H2′β, 6′β), 3.50 (4H, d, J=5.8 Hz, H1), 4.50–4.82 (4H, m, H2′α, 6′α), 7.36 (1H, dm, J=7.9 Hz, H5‴), 7.41–7.47 (2H, m, J=7.6 Hz, H4‴, 6‴) 7.52–7.54 (1H, m, H2‴). 13C-NMR (CDCl3) δ: 28.50 (C3′, 5′), 39.03 (C4′), 43.37 (C2′, 6′), 67.55 (C1), 119.61 (q, J=4.1 Hz, C2‴), 121.40 (q, J=4.1 Hz, C4‴), 123.82 (q, J=272.6 Hz, CF3), 125.19 (C6‴), 129.35 (C5‴), 131.15 (q, J=32.4 Hz, C3‴), 152.82 (C1‴), 165.78 (C2″, 4″), 170.60 (C6″). Positive-ion FAB-MS m/z: 468 (M+H+). HR-FAB-MS m/z: 468.2229 (Calcd for C22H29F3N5O3: 468.2222). Anal. Calcd for C22H28F3N5O3 : C, 56.52; H, 6.04; N, 14.98. Found: C, 56.52; H, 5.90; N, 14.99.
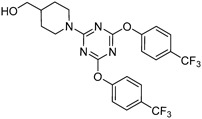
[1-[4,6-Bis[3-(trifluoromethyl)phenoxy)]-1,3,5-triazin-2-yl]piperidin-4-yl]methanol (6kkp)This compound was prepared from phenol (kH) and amine (pH) by using Method C at room temperature for 1 h in Step 1 and then at room temperature for 18 h in Step 2 with the ratio of TCT AZ : kH : collidine : pH=1 : 2.4 : 2.4 : 2. Purification of products by flash chromatography (n-hexane : EtOAc=85 : 15→70 : 30) gave 6kkp (69%) and 5kpp (29%). An analytical sample of 6kkp was obtained by recrystallization from diisopropyl ether as colorless crystals.
6kkp: mp 135.0–136.5°C (from diisopropyl ether). IR (KBr) cm−1: 1596 (C=N), 1326 (C–F), 1123, 1068 (C–O). 1H-NMR (CDCl3) δ: 1.12–1.22 (4H, m, H3′β, 5′β), 1.53 (1H, br s, OH), 1.74–1.80 (3H, m, H4′α, 3′α, 5′α), 2.84 (2H, dt, J=13.4, 2.4 Hz, H2′β, 6′β), 3.51 (2H, d, J=5.8 Hz, H1), 4.57 (2H, dm, J=13.4 Hz, H2′α, 6′α), 7.30–7.34 (2H, m, H5‴), 7.43–7.48 (6H, m, H2‴, 4‴, 6‴). 13C-NMR (CDCl3) δ: 28.37 (C3′, 5′), 38.71 (C4′), 43.88 (C2′, 6′), 67.19 (C1), 119.20 (q, J=4.1 Hz, C2‴), 122.19 (q, J=4.1 Hz, C4‴), 123.58 (q, J=273.1 Hz, CF3), 125.15 (C6‴), 129.74 (C5‴), 131.65 (q, J=32.8 Hz, C3‴), 152.12 (C1‴), 166.21 (C2″, 4″), 171.89 (C6″). Positive-ion FAB-MS m/z: 515 (M+H+). HR-FAB-MS m/z: 515.1524 (Calcd for C23H21F6N4O3: 515.1518). Anal. Calcd for C23H20F6N4O3: C, 53.70; H, 3.92; N, 10.89. Found: C, 53.63; H, 3.76; N, 10.86.
1,1′-[6-(4-Trifluoromethyl)pheoxy)-1,3,5-triazine-2,4-diyl]bis-4-piperidinemethanol (5lpp) and [1-[4,6-Bis[4-(trifluoromethyl)phenoxy]-1,3,5-triazin-2-yl]piperidin-4-yl]methanol (6llp)These compounds were prepared from phenol (lH) and amine (pH) by Method C at room temperature for 1 h in Step 1 and then at room temperature for 0.5 h in Step 2 with the ratio of TCT AZ : lH : collidine : pH=1 : 1.8 : 1.8 : 2. Purification of products by flash chromatography (CH2Cl2 : EtOH=97 : 3→93 : 7) gave 6llp (73%) and 5lpp (19%). Analytical samples of 5lpp and 6llp were obtained as colorless crystals by recrystallization from EtCN and EtOH, respectively.
5lpp: 162–164°C (from EtCN). IR (KBr) cm−1: 1583, 1527 (C=N), 1328 (C–F), 1064, 1038 (C–O). 1H-NMR (CDCl3) δ: 1.10–1.25 (4H, m, H3′β, 5′β), 1.44 (2H, br s, OH), 1.72–1.86 (6H, m, H4′α, 3′α, 5′α), 2.74–2.87 (4H, m, H2′β, 6′β), 3.51 (4H, d, J=6.1 Hz, H1), 4.50–4.86 (4H, m, H2′α, 6′α), 7.30 (2H, d, J=8.4 Hz, H2‴, 6‴), 7.61 (2H, d, J=8.4 Hz, H3‴, 5‴). 13C-NMR (CDCl3) δ: 28.53 (C3′, 5′), 39.04 (C4′), 43.39 (C2′, 6′), 67.57 (C1), 122.28 (C2‴, 6‴), 124.17 (q, J=33.1 Hz, CF3), 126.16 (q, J=3.1 Hz, C3‴, 5‴), 126.77 (q, J=33.1 Hz, C4‴), 155.38 (C6″), 165.82 (C2′, 4″), 170.57 (C1″). Positive-ion FAB-MS m/z: 468 (M+H+). HR-FAB-MS m/z: 468.2226 (Calcd for C22H29F3N5O3: 468.2222). Anal. Calcd for C22H28F3N5O3: C, 56.52; H, 6.04; N, 14.98. Found: C, 56.44; H, 5.93; N, 14.99.
6llp: mp 196–199°C (from EtOH). IR (KBr) cm−1: 1562, 1540 (C=N), 1329 (C–F), 1060, 1047 (C–O). 1H-NMR (CDCl3) δ: 1.14–1.24 (2H, m, H3′β, 5′β), 1.47 (1H, br s, OH), 1.73–1.84 (3H, m, H4′α, 3′α, 5′α), 2.85 (2H, dt, J=11.6, 2.1 Hz, H2′β, 6′β), 3.51 (2H, d, J=5.8 Hz, H1), 4.59 (2H, dm, H2′α, 6′α), 7.45 (4H, d, J=5.8 Hz, H2‴, 6‴), 7.61 (4H, d, J=8.5 Hz, H3‴, 5‴). 13C-NMR (CDCl3) δ: 28.41 (C3′, 5′), 39.69 (C4′), 43.91 (C2′, 6′), 67.19 (C1), 122.20 (C2‴, 6‴), 123.91 (q, J=272.1 Hz, CF3), 126.54 (q, J=4.1 Hz, C3‴, 5‴), 127.82 (q, J=33.1 Hz, C4‴), 154.53 (C4″, 6″), 166.32 (C2″), 171.80 (C1‴). Positive-ion FAB-MS m/z: 515 (M+H+). HR-FAB-MS m/z: 515.1523 (Calcd for C23H21F6N4O3: 515.1518). Anal. Calcd for C23H20F6N4O3: C, 53.70; H, 3.92; N, 10.89. Found: C, 53.77; H, 4.06; N, 10.81.
2,4-Bis(heptyloxy)-6-isopropoxy-1,3,5-triazine (4dde) and 2,4-Bis(cyclopentyloxy)-6-isopropoxy-1,3,5-triazine (4egg)The intermediate 2e for the preparation of compounds 4dde and 4egg was prepared from the reaction of compound 1 and 2-PrOH (eH) by using Method C at room temperature for 2.5 h with the ratio of TCT AZ : eH : collidine=1 : 2.6 : 2. After removal of the precipitated material (collidine·HCl), purification of the obtained crude product by open column chromatography (n-hexane : EtOAc=90 : 10) gave 2e8) (83%). Title two compounds were prepared from the reactions of compound 2e with alcohol dH or gH by using Method A under the same conditions as those for the preparation of 4e (Entry 4 in Table 1) with the ratio of 2e : dH or gH : NaH=1 : 4 : 2. Separation of the products by flash chromatography (n-hexane : EtOAc=95 : 5→93 : 7) gave 4dde (75%) as colorless oil or 4egg (42%) as a white solid, respectively. Recrystallization from 2-PrOH–H2O gave 4egg as white crystals.
4dde: IR (NaCl) cm−1: 1560 (C=N), 1146, 1102 (C–O). 1H-NMR (CDCl3) δ: 0.88 (6H, t, J=7.0 Hz, H7′), 1.26–1.36 (12H, m, H4′, 5′, 6′), 1.38 (6H, d, J=6.1 Hz, H1″, 3″), 1.40–1.46 (4H, m, H3′), 1.78 (4H, qu, J=6.7 Hz, H2′), 4.37 (4H, t, J=6.7 Hz, H1′), 5.36 (1H, qu, J=6.1 Hz, H2″). 13C-NMR (CDCl3) δ: 13.92 (C7′), 21.69 (C1″, 3″), 22.47 (C6′), 25.71 (C3′), 28.60 (C2′), 28.86 (C4′), 31.64 (C5′), 68.31 (C1′), 71.43 (C2″), 172.52 (C6), 173.14 (C2, 4). Positive-ion FAB-MS m/z: 368 (M+H+). HR-FAB-MS m/z: 368.2909 (Calcd for C20H38N3O3: 368.2913). Anal. Calcd for C20H37N3O·0.2H2O: C, 64.73; H, 10.16; N, 11.32. Found: C, 64.71; H, 10.15; N, 11.43.
4egg: mp 94–97°C (from 2-PrOH–H2O). IR (KBr) cm−1: 1562 (C=N), 1133, 1101 (C–O). 1H-NMR (CDCl3) δ: 1.37 (6H, d, J=6.1 Hz, H1″, 3″), 1.56–1.65 (4H, m, H3′, 4′), 1.78–1.89 (8H, m, H2′, 3′, 4′, 5′), 1.92–2.00 (4H, m, H2′, 5′), 5.34 (1H, qu, J=6.1 Hz, H2″), 5.46 (2H, qu, J=3.1 Hz, H1′). 13C-NMR (CDCl3) δ: 21.79 (C1″, 3″), 23.79 (C3′, 4′), 32.71 (C2′, 5′), 71.27 (C2″), 80.61 (C1′), 172.48 (C6), 172.80 (C2, 4). Positive-ion FAB-MS m/z: 308 (M+H+). HR-FAB-MS m/z: 308.1977 (Calcd for C16H26N3O3: 308.1974). Anal. Calcd for C16H25N3O3: C, 62.52; H, 8.20; N, 13.67. Found: C, 62.57; H, 8.22; N, 13.55.
Antiviral Activity Assay and Cytotoxicity of Synthesized Trisubstituted TAZ DerivativesThe anti-HSV-1 activities (EC50) of the synthesized TAZ derivatives were measured by using a plaque reduction assay22) and their cytotoxicity against Vero cells (IC50) was also evaluated. The results are summarized in Table 2 together with data for aciclovir.23) Calculated log P values24) for the compounds are also shown in Table 2. There were few distinct correlations between log P values and EC50 values or between log P values and IC50 values among the compounds listed in Table 2.
Calorimetric ExperimentsThe heat of binding between a tripodal receptor-type TAZ derivative 4e and sugars was measured in aqueous 25% 2-propanol solution at 298.15 K by using an isothermal titration calorimeter (Thermal Activity Monitor 2270). Titrations were performed by stepwise injection of a sugar-containing solution (3.5–4.6 mM, or ca. 5 mg/mL of dermatan sulfate or heparin sulfate) into a reaction cell (3 mL) loaded with the compound 4e solution (ca. 310–460 µM). Many commercial sugar derivatives of monosaccharide, disaccharide, trisaccharide, tetrasaccharide, oligosaccharide, aminosugar and some sulfated glycosaminoglycans were used for calorimetric experiments; however, observed reactions of compound 4e with many sugar derivatives were not exothermic. Three typical examples of exothermic binding reactions for methyl esters of monosaccharide (MeO-α/β-Gal and MeO-α/β-Man) are shown in Table 3. The data obtained were analyzed by NanoAnalyze™ Software (TA Instruments, U.S.A). Binding stoichiometry, Ka and ΔH are shown in Table 3. The values of ΔG and −TΔS were also calculated from the equation ΔG=−RT ln Ka=ΔH−TΔS (where R is the gas constant and T is absolute temperature). The heat of binding between a tripodal receptor-type TAZ derivative 4g and MeO-α/β-Gal was also measured in aqueous 25% 2-propanol solution at 298.15 K.