Results and Discussion
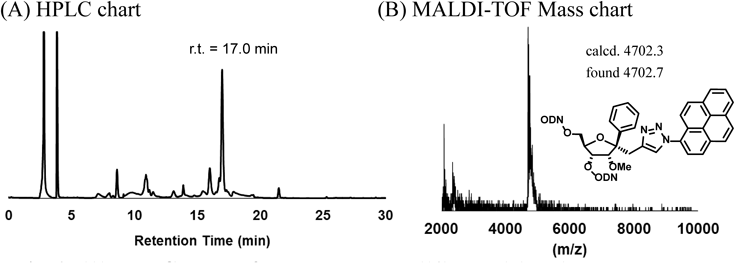
Chart 1 depicts the synthesis of 14-mer TFOs containing a variety of molecules. (1S,2R,3R,4R)-1-(2-Formylethyl)-2-O-methyl-1-phenyl-3,5-O-tetraisopropyldisiloxyribose (11)14) was synthesized as described in our previous paper. The aldehyde group was converted to the alkyne unit using Ohira–Bestmann reagents, followed by treatment with tetrabutylammonium fluoride (TBAF) to obtain the diol compound (12). This diol compound was further transformed into the amidite compound (13). The amidite unit 13 was applied to an automated DNA/RNA solid phase synthesizer and incorporated into intra-oligodeoxynucleotides (intra-ODNs) targeting the survivin oncogene.19,23) Then, the synthesized ODNs were reacted with corresponding azido-modified compounds (1–10)24–29) under the Huisgen cycloaddition reaction conditions30) on controlled pore glass (CPG). The CPG resins were washed with water and acetonitrile in succession. For example, during the purification of TFOs, the synthesized ODNs containing pyrene (4,4′-dimethoxytrityl (DMTr)-TFO(10)) were cleaved from CPG and the protecting groups were removed in a solution containing 28% ammonia at 55°C for 5 h, followed by purification by HPLC (Fig. 2A). After removing the DMTr group, matrix assisted laser desorption/ionization-time of flight (MALDI-TOF)/MS measurements confirmed that the modified TFO(10) was obtained, as shown in Fig. 2B. All synthesized TFOs were treated in the same way as described for TFO(10). The HPLC retention times and MALDI-TOF/MS data of the modified TFOs are summarized in Table 1. Simultaneously, TFO(propargyl) and TFO(ethyl(T)) were synthesized and purified as control TFOs (Table 1).

Chart 1
The ability of the synthesized TFOs to form triplexes was evaluated by a gel shift assay using 100 nM FAM-labeled target duplexes in 20 mM Tris–HCl, 20 mM MgCl2, 2.5 mM spermidine, 10% sucrose, pH 7.5. Triplex formation was identified by the presence of lower mobility bands in the 10% non-denatured polyacrylamide gel (Fig. 3). These bands were then quantified, and the equilibrium association constant (Ks) was calculated for each TFO (Table 2).
Table 2. Equilibrium Association Constant (
Ks)
a)| TFO | Ks (106 M−1) | TFO | Ks (106 M−1) | TFO | Ks (106 M−1) |
|---|
| 17-mer TFO(T) | 2.02 | 14-mer TFO(G) | n.d.b) | TFO(6) | n.d.b) |
| 17-mer TFO(A) | 1.00 | 14-mer TFO(C) | n.d.b) | TFO(7) | n.d.b) |
| 17-mer TFO(G) | 0.94 | TFO(1) | n.d.b) | TFO(8) | n.d.b) |
| 17-mer TFO(C) | 0.07 | TFO(2) | n.d.b) | TFO(9) | n.d.b) |
| | TFO(3) | n.d.b) | TFO(10) | 1.08 |
| 14-mer TFO(T) | n.d.b) | TFO(4) | n.d.b) | TFO(propargyl) | n.d.b) |
| 14-mer TFO(A) | n.d.b) | TFO(5) | n.d.b) | TFO(ethyl(T)) | n.d.b) |
a) Triplex formation was evaluated under the same conditions as described in the footnote to Fig. 3. Ks (106 M−1)=[Triplex]/[Duplex][TFO], errors within 10%. b) n.d.: not detected under the above-mentioned conditions.
Although the natural 17-mer TFO(T) was able to form triplex DNA, the natural 14-mer TFO(T) did not exhibit bands representing triplex DNA for this target site under the assay conditions (Fig. 3). TFO(propargyl), triazole modified TFO(1), alkyl modified TFO(2 and 3), polyether modified TFO(4 and 5), polyamine modified TFO(6 and 7), phenyl modified TFO(8) and naphthalene modified TFO(9) did not exhibit significant stabilizing effects on the formation of triplex DNA. Among these modifications, only pyrene modified TFO(10) showed triplex formation with target duplex DNA. Surprisingly, no triplex band was observed for TFO(ethyl(T)), therefore, we thought that the 14-mer TFO with the WNA derivative12–18) would not form the triplex DNA for the region including one CG site. The results of the gel-shift assay and the Ks value demonstrated that pyrene contributed to the stability of triplex formation with target duplex DNA.
To investigate the interaction of this pyrene-modified TFO with duplex DNA, we measured the fluorescence of TFO(10) before and after the addition of the duplex DNA. The fluorescence of the pyrene was gradually quenched when the target duplex DNA was added (Fig. 4A). In contrast, when duplex DNA without the target site was added, no quenching effect was observed (Fig. 4B). These results indicate that the pyrene molecule intercalated into duplex DNA, assisting in the formation of triplex DNA.31) Computational chemistry was used to predict the effect of this pyrene. The molecular modeling result is presented in Fig. 5, with TFO presented with CPK and the artificial nucleoside analogue with pyrene depicted in green. This result demonstrates that the pyrene is intercalated between the base pair of the duplex DNA. Accordingly, we investigated the stabilizing effect of the pyrene derivative on the different base pairs, GC, AT or TA base pairs (Fig. 6). Although the triplexes were not stable, the pyrene modified TFO(10) showed a stabilizing effect for all base pairs. These results have supported the intercalation effect of the pyrene part.
The pyrene modified 14-mer (TFO(10), the 17-mer TFO(T)), other TFOs (TFO(T), TFO(ethyl(T)), and a 14-mer scrambled sequence (5′-GGA GGA GGA GGA TA-3′) as a control were transfected into A549 (human lung adenocarcinoma epithelial cell line) cells using Oligofectamine™ and PLUS reagent. The cells were cultured for 72 h, and then cell viability was evaluated by CellTiter 96®.19) These results are presented in Fig. 7 as a bar graph. The cell viabilities decreased as the TFO concentration increased. Comparing the 14-mer natural TFO(T) with the 17-mer natural TFO(T) demonstrates that longer TFO sequences enhance the anti-proliferative effect. Unfortunately, the pyrene modified TFO(10) inhibited cell proliferation to the same degree as the natural TFO(T). This result suggests that these TFOs may be degraded by nucleases in the cultured cells because of the lack of modifications at the end of the ODNs.
In summary, this paper reports the successful synthesis of (1S,2R,3R,4R)-3-O-[2-cyanoethoxy(diisopropylamino)phosphino]-5-O-dimethosytrityl-2-O-methyl-1-phenyl-1-propargylribose (13) as a novel platform molecule for the synthesis of TFOs that can recognize duplex DNA including the conversion base pair site. Among the various TFOs synthesized here, the pyrene-modified TFO(10) exhibited the best stabilization of the formation of triplex DNA where the target duplex DNA includes one CG site, and inhibited cell proliferation of A549 cells. This study is valuable for the development of gene targeting using artificial triplex-forming oligonucleotides (TFOs). In the future work, to improve the anti-proliferative effects of these TFOs in living cells, 3′- and/or 5′-terminal modification will be introduced to TFOs to limit degradation.
Experimental
The 1H-NMR (400 MHz) and 13C-NMR (125 MHz) spectra were recorded by Varian UNITY-400 and INOVA-500 spectrometers. The 31P-NMR (161 MHz) spectrum was recorded using 10% phosphoric acid in D2O as the internal standard at 0 ppm. The infrared (IR) spectra were obtained using a Perkin-Elmer FTIR-SpectrumOne. The high-resolution mass spectra were recorded by an Applied Biosystems Mariner System 5299 spectrometer using bradykinin, angiotensin and neurotensin as the internal standards. DNA was synthesized by a Nihon Techno Service NTS-H6 DNA/RNA synthesizer and purified by HPLC. The MALDI-TOF/MS spectra were recorded by a BRUKER DALTONICS Microflex. The UV-Vis spectra and fluorescence spectra were measured by Beckman-Coulter DU-800 and JASCO FP-6500 instruments, respectively. Natural DNA was purchased from Genenet Co., Ltd. and Gene Design, Inc., Japan.
(1S,2R,3R,4R)-2-O-Methyl-1-phenyl-1-propargylribose (12)Under an Ar atmosphere, the solution of compound 11 (600 mg, 1.18 mmol)14) in dry methanol (3.0 mL) was added to a mixture of dimethyl(1-diazo-2-oxopropyl)phosphonate (670 mg, 3.55 mmol) and potassium carbonate (280 mg, 2.00 mmol) in dry methanol (6.0 mL) at 0°C. After stirring for 3 h at the same temperature, the reaction was quenched with water and extracted with diethylether. The organic layer was washed with saturated sodium chloride solution and dried over Na2SO4, and then the solvent was removed under vacuum. The residue was purified by silica gel column chromatography (Kanto 60N, hexane–EtOAc=10 : 1), yielding a colorless oil (360 mg, 0.72 mmol, 61%). This oil compound (325 mg, 0.64 mmol) was dissolved in dry tetrahydrofuran (THF) (6.4 mL) and treated with tetrabutyl ammonium fluoride in THF (1 M, 1.35 mL, 1.35 mmol). After stirring for 2 h, the solvent was removed under vacuum. The residue was purified by silica gel column chromatography (Kanto 60N, hexane–EtOAc=1 : 1), yielding the colorless oil 12 (167 mg, 0.64 mmol, 99%). IR (cm−1): 3404, 2939, 1447. 1H-NMR (400 MHz, CDCl3) δ (ppm): 7.48 (2H, d, J=7.02 Hz), 7.35 (2H, t, J=7.02 Hz), 7.27 (1H, t, J=7.02 Hz), 4.17–4.13 (2H, m), 3.90–3.86 (2H, m), 3.71–3.67 (1H, m), 3.61 (3H, s), 2.93 (1H, dd, J=14.0, 2.8 Hz), 2.82 (1H, dd, J=14.0, 2.8 Hz), 2.11 (2H, br s), 1.87 (1H, t, J=2.8 Hz). 13C-NMR (125 MHz, CDCl3) δ (ppm): 143.7, 128.3, 127.5, 124.7, 87.6, 85.9, 84.1, 80.6, 70.9, 70.6, 63.2, 60.3, 27.2. High resolution-electrospray ionization (HR-ESI)-MS m/z: Calcd for (M+Na)+ C15H18O4Na, 285.1097; Found, 285.1051.
(1S,2R,3R,4R)-3-O-[2-Cyanoethoxy(diisopropylamino)phosphino]-5-O-dimethosytrityl-2-O-methyl-1-phenyl-1-propargylribose (13)Under an Ar atmosphere, compound 12 (71 mg, 0.27 mmol) was dissolved in pyridine (5.4 mL), followed by the addition of DMTrCl (139 mg, 0.41 mmol). After stirring for 90 min at room temperature, the reaction mixture was quenched with water, and then extracted with AcOEt. The organic layer was successively washed with brine, dried over Na2SO4, and evaporated under vacuum to obtain a pale yellow oil. The resulting oil was purified by silica gel column chromatography (Kanto 60N, hexane–EtOAc=5 : 1 to 2 : 1) to obtain a colorless foam (152 mg, 0.27 mmol). To a solution of this compound (100 mg, 0.18 mmol) in CH2Cl2 (3.5 mL), diisopropylethylamine (0.19 mL, 1.06 mmol) and 2-cyanoethyl-N,N-diisopropylchlorophosphoramidite (0.13 mL, 0.531 mmol) were added at 0°C, and the reaction mixture was stirred at 0°C for 3 h. The reaction mixture was quenched with a saturated NaHCO3 solution and extracted with EtOAc. The organic layer was dried over Na2SO4 and then evaporated under vacuum to obtain a colorless oil. The oil was purified by flash column chromatography (Fuji Silysia FL 60D, hexane–EtOAc=5 : 1 to 3 : 1) to obtain a colorless foam. This foam was precipitated again by hexane at −78°C, yielding 13 as a white foam (100 mg, 0.13 mmol, 74%). IR (cm−1): 2913, 1770. 1H-NMR (400 MHz, CDCl3) δ (ppm): 7.66–7.61 (2H, m), 7.34–7.30 (3H, m), 7.27–7.14 (9H, m), 6.78–6.72 (4H, m), 4.39–4.34 (0.8H, m), 4.32–4.26 (1.2H, m), 3.93 (0.4H, d, J=5.19 Hz), 3.87–3.83 (1.6H, m), 3.76 (2H, s), 3.75 (4H, s), 3.57–3.53 (3H, m), 3.48 (2H, s), 3.42 (1H, s), 3.39–3.38 (0.4H, m), 3.15–3.10 (2H, m), 2.89–2.85 (1.6H, m), 2.58 (1.6H, t, J=6.10 Hz), 2.21 (0.4H, t, J=6.72 Hz), 1.81 (1H, t, J=2.74 Hz), 1.18 (2.8H, t, J=6.72 Hz), 1.14 (5.2H, t, J=6.72 Hz), 0.99 (4H, t, J=7.02 Hz). 31P-NMR (161 MHz, CDCl3) δ (ppm): 150.65, 149.61. ESI-MS [M+H]+ 765.4. HR-ESI-MS m/z: Calcd for (M+H)+ C45H54N2O7P, 765.3663; Found, 765.3668.
Synthesis of Triplex Forming Oligonucleotides (TFOs) Containing Artificial Nucleoside AnaloguesThe TFOs were synthesized at a 1.0 µmol scale by a NTS-H6 DNA RNA synthesizer (Nihon Techno Service Co., Ltd.) using the standard protocol. After synthesis of the TFOs containing the platform molecule 12, the CPG was treated with the appropriate azide-modified compound under the Cu(I)-catalyzed azide-alkyne cycloaddition conditions. A portion of the CPG beads were treated with Cu(II) sulfate (0.1 M of aqueous solution, 50 µL), sodium ascorbate (0.1 M in aqueous solution, 50 µL), tris-(benzyltriazolylmethyl)amine (TBTA, 0.1 M of dimethyl sulfoxide (DMSO) solution, 100 µL) and corresponding azide-compound (0.1 M of DMSO solution, 250 µL) at room temperature for 2 h. The CPG beads were successively washed with water and CH3CN, and suspended in a 28% ammonia solution at 55°C for 5 h. After filtration, the mixture was purified by reversed phase HPLC (ODS column, linear gradient between 0.1 M triethylammonium acetate (TEAA) buffer and CH3CN), and the DMTr-protecting group was removed by 5% acetic acid. The structures of the synthesized TFOs were confirmed by MALDI-TOF mass measurements, which were summarized in Table 1 in the manuscript.
Gel-Shift AssayThe 5′ FAM-labeled target oligonucleotides were purchased from Genenet Co., Ltd. The targeted duplex DNAs (100 nM) were added to different concentrations of the corresponding TFOs (from 10 to 1000 nM) in 20 mM Tris–HCl, 5 mM MgCl2, 2.5 mM spermidine and 10% sucrose, pH 7.5. These mixtures were incubated at 30°C for 12 h, and electrophoresis was carried out at 10°C using a 10% non-denatured polyacrylamide gel. The fluorescent bands were detected by an Image Quant LAS-4000 image analyzer, and the intensities of these bands were quantified to calculate the equilibrium association constant (Ks). The Ks values for triplicate experiments were averaged, and summarized in Table 2. Errors were estimated to be within 10%.
Molecular ModelingAn initial triplex structure was generated using the solution structure of PDB134D by converting the natural nucleoside in the middle of TFO to the β-phenyl and α-triazole-pyrene modified nucleoside analogue. The initial complex structure was optimized by the MacroModel using an OPLS_2005 force field in GB/SA solvation model of water. The minimization was performed using PRCG method until the gradient reached to 0.05 kJ/mol·Å.
Cell Proliferation AssayCells were treated with TFOs and transfection reagents for 4 h and then incubated for 72 h at 37°C and 5% CO2, following the protocol described in a previous paper. After 72 h incubation, 15 µL of the CellTiter 96® reagent (Promega) was added to each well. After 2 h incubation, the absorbance was measured at 490 nm using a microplate reader. The values of the cell viability were calculated from three replicate experiments, and each experiment was carried out three or more times.