Abstract
An attempt to synthesize aglycone 1 derived from 2,3,5,4′-tetrahydroxystilbene-2-O-β-glucoside (THSG) via the Wittig reaction and Mizoroki–Heck reaction is described. In the Wittig protocol, 2,3,5,4′-tetramethoxystilbene 2 was obtained. Additionally, a palladium-catalyzed Mizoroki–Heck reaction strategy yielded 2-aryl-2,3-dihydrobenzofuran 13 instead of derivative 12 in good yield.
Dietary polyphenols, such as flavonoids,1) stilbenes2) and lignans,3) are now recognized for their beneficial uses in human health care, such as in the treatment and prevention of cardiovascular diseases. Cancer prevention has also been associated with consumption of a diet rich in these polyphenols.4,5)
In particular, resveratrol (3,5,4′-trihydroxy-trans stilbene), a polyphenolic natural product present as a glycoside6) or methoxide,7) has chemoprevention effects against cardiovascular diseases,8) cancer,9) Alzheimer’s disease and aging10,11) (Fig. 1).
However, the therapeutic potential of resveratrol is limited because free resveratrol is rapidly absorbed in the upper GI tract following oral dosing, quickly metabolized to glucuronides and sulfates and quickly cleared from the circulation.12)
2,3,5,4′-Tetrahydroxystilbene-2-O-β-D-glucoside (THSG) is also a polyphenolic glycoside extracted from the dried root tuber of Polygonum multiflorum (Fig. 1).
The dried root tuber of Polygonum multiflorum (Radix Polygoni mutiflori), a traditional herbal drugs in the Chinese pharmacopeia, has been used as an important health protective agent and nutritional supplement for thousands of years.13)
Recent pharmacological studies indicated that THSG has strong antioxidative and free radical scavenging activity.14–16) In addition, THSG is able to reduce hyperlipidemia, prevent lipid peroxidation, and protect the cardiovascular system.17) It is also effective in the prophylaxis and therapeutic treatment of Alzheimer’s disease.18)
The usefulness of THSG, however, is limited by its instability upon exposure to irradiation and oxygen or at a particular pH and temperature.19,20)
The bioactivities of polyhydroxy stilbene compounds have mainly been attributed to their antioxidant property, which is related to the position of hydroxyl groups.2,10)
In addition, polyhydroxy stilbenes with catechol substituents dramatically increase antioxidative activity, but the glycosylation of stilbene reduces their activity compared to the corresponding aglycone.6,21,22)
Therefore, the aglycone 1 (2,3,5,4′-tetrahydroxystilbene) from THSG is a useful antioxidant and free radical scavenger and possesses potential therapeutic application to human disease.
To the best of our knowledge, the aglycone 1 derived from THSG has rarely been reported on in the literature, and only two reports mention antioxidative activities. Reports indicate that aglycone 1 has very high 1,1-diphenyl-2-picrylhydrazyl (DPPH) radical scavenging activity (IC50=0.38 µM) in vitro, which is nearly a hundred times that of THSG (IC50=40 µM).23,24)
Our interest in the aglycone 1 stemmed from its high antioxidative activity, therapeutic potential and novel mechanism of action. However, the systematic investigation of biological and therapeutic properties of the aglycone 1 is somewhat limited by its general accessibility. Besides methods of aglycone 1 extraction from plants, efficient methods of aglycone 1 synthesis would be highly desirable.
Results and Discussion
Firstly, we tried to prepare the aglycone 1 via hydrolysis with β-glucosidase according to a previously reported procedure23,24) (Chart 1).

Chart 1. Hydrolysis of THSG with β-Glucosidase
However, the desired aglycone 1 was not observed under the conditions. We also tried various other methods and found that glucose was among the reaction products of hydrolysis of THSG with β-glucosidase, indicating the occurrence of deglucosylation. However, the presence of the aglycone 1 could not be confirmed in the obtained/resulting products by HPLC or 1H-NMR.
Next, we decided to investigate the synthesis of the aglycone 1 through the Wittig reaction followed by deprotection of (E)-2,3,5,4′-tetramethoxystilbene (2) (Chart 2).
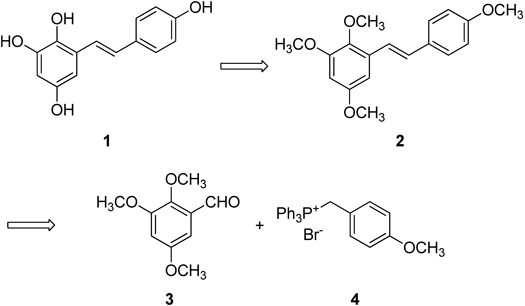
Chart 2. Synthetic Plan of 1 through Wittig Reaction
The (E)-2,3,5,4′-tetramethoxystilbene (2) was synthesized in eight steps beginning with Vanillin. The 2,3,5-trimethoxybenzaldehyde (3), which was not available commercially, was synthesized as previously described25,26) (Chart 3).
Vanillin is regioselectively brominated at the 5-position to give arylbromide 5 in good yield (90%). Subsequent O-methylation using methyl iodide was followed by Dakin oxidation using m-chloroperbenzoic acid in refluxing CH2Cl2 of the resulting product to phenol 7, which underwent nitrile- for -Br exchange on treatment with CuCN in refluxing N,N-dimethylformamide (DMF) in good yield (89%). After conversion of the OH group into methyl ether 9, reduction of the nitrile functional group with diisobutylaluminium hydride followed by mild acidic workup afforded quantitatively the corresponding aldehyde 3.
The 2,3,5,4′-tetramethoxystilbene (2) was prepared by the Wittig reaction. 4-Methoxybenzyltriphenylphosphonium bromide was synthesized from 4-methoxybenzylalcohol by bromination, followed by reaction with triphenylphosphine.27)
The phosphonium salt was reacted with the aldehyde 3 in the presence of 15% NaOH as base and nBu4NI to give the desired 2,3,5,4′-tetramethoxystilbene as a mixture of E and Z isomers (Chart 4).

Chart 4. Synthesis of 2
However, chromatographic separation of the E and Z isomers was very difficult in our hands. Fortunately, Z to E isomerization was easily accomplished under iodine catalysis. That is to say, the E/Z mixture of 2,3,5,4′-tetramethoxystilbene was quantitatively converted into the corresponding E stereoisomer by treatment with a catalytic amount of iodine in hexane at reflux.28)
Next, we attempted to convert 2,3,5,4′-tetramethoxystilbene 2 directly into the aglycone 1 by deprotection of methyl ether. Boron tribromide is known to demethylate aromatic methyl ether. Treatment of (E)-2,3,5,4′-tetramethoxystilbene (2) with boron tribromide gave an intractable brown solid that could not be characterized. Furthermore, demethylation of aromatic methyl ether with trimethysilyl iodide and pyridine in CH2Cl2 at −78°C or with sodium isopropyl thiolate in DMF at reflux did not give the aglycone 1.29)
This appeared to confirm the sensitivity of phenolic functional groups such as those in THSG to these reaction conditions. The presence of the second hydroxyl group in the ortho or para position is speculated to be due to its isomerization to o-quinone or p-quinone derivatives. THSG is also thought to be oxidized and hydrolyzed, ultimately, resulting in degradative decomposition, but details of the underlying mechanism remain unknown.19) Despite the satisfactory results obtained regarding synthesis of (E)-2,3,5,4′-tetramethoxystilbene (2), the Wittig protocol has general limitations. This method applied to the synthesis of the aglycone 1 requires a preliminary protection of phenolic groups of the starting aromatic compounds and the deprotection of these protective groups under alternative milder conditions.
Because of these difficulties we had to change our synthetic approach as outlined in Chart 5. The key step in the synthesis of the aglycone 1 is to form the stilbene via the Mizoroki–Heck reaction.29) A palladium-catalyzed Mizoroki–Heck reaction has been also used previously to form resveratrol, glucopyranosides of resveratorol and polymethoxystilbenes.2,30,31)

Chart 5. Mizoroki–Heck Reaction
In our strategy, we used the Mizoroki–Heck reaction of a suitable bromo derivative 10 with styrene derivative 11 (Chart 5). We chose to utilize the acetyl derivative 11 because the Mizoroki–Heck reaction is known to give a better yield. The 2-bromo-6-methoxy-1,4-hydroquinone (10), was obtained by the Dakin reaction of 5-bromovanillin with m-chloroperbenzoic acid followed by hydrolysis of the ester group. The Mizoroki–Heck reaction was carried out using 5 mol% of Pd(OAc)2 as catalyst, (the concentration of which was calculated based upon 10), Na2CO3 as base, nBu4NI as phase transfer catalyst, and DMF as solvent. However, this coupling reaction did not afford the desired product 12 but instead 2-aryl-2,3-dihydrobenzofuran derivative 13 in good yield (87%).32,33)
This reaction appears to proceed as depicted in Chart 6: oxidative addition of the hydroquinone 10 to Pd(0) and subsequent addition of the resulting arylpalladium complex to the carbon–carbon double bond of styrene derivative 11.34)
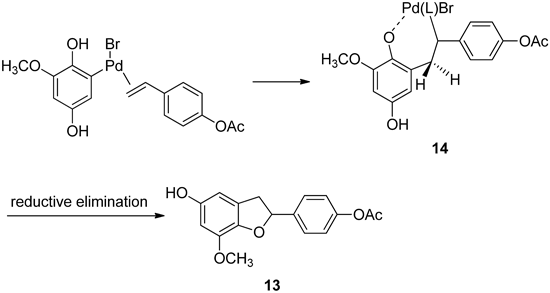
Chart 6. Possible Mechanism of Mizoroki–Heck Reaction
Subsequent intramolecular nucleophilic attack of the phenoxy group (2-hydroxyl group) on the σ-alkylpalladium intermediate (14) results in the observed dihydrobenzofuran and in regeneration of the Pd(0) catalyst. The high yield of dihydrobenzofuran can be probably accounted for by assuming that coordination of the phenolic (or phenoxy) group to the palladium in the σ-alkylpalladium intermediate (14) makes it difficult to achieve the conformation required for syn β-hydride elimination of one of the benzilic hydrogens. Thus reductive elimination of the hydride-palladium species is directed towards the 2-aryl-2,3-dihydrobenzofuran (13) by coordination of the phenolic (or phenoxy) oxygen nucleophile to the palladium in the 14.
Conclusion
We are currently exploring the application of the palladium-catalyzed Mizoroki–Heck reaction to the synthesis of 2-aryl-2,3-dihydrobenzofuran derivatives, although we could not afford the aglycone 1 (2,3,5,4′-tetrahydroxystilbene). It is suggested that it is necessary to protect the 2-hydroxyl group for achieve to synthesis of aglycone 1. The results of these studies will be reported in due course.
Conflict of Interest
The authors declare no conflict of interest.
References
- 1) Agati G., Azzarello E., Pollastri S., Tattini M., Plant Sci., 196, 67–76 (2012).
- 2) Albert S., Horbach R., Deising H. B., Siewert B., Csuk R., Bioorg. Med. Chem., 19, 5155–5166 (2011).
- 3) Saleem M., Kim H. J., Ali M. S., Lee Y. S., Nat. Prod. Rep., 22, 696–716 (2005).
- 4) Bharti R., Ahuja G., Ganapathy S. P. S., Dakappa S. S., J. Pharm. Res., 5, 4278–4287 (2012).
- 5) Hur S. J., Lee S. Y., Kim Y.-C., Choi I., Kim G.-B., Food Chem., 160, 346–356 (2014).
- 6) Fabris S., Momo F., Ravagnan G., Stevanato R., Biophys. Chem., 135, 76–83 (2008).
- 7) Regev-Shoshani G., Shoseyov O., Kerem Z., Biochem. Biophys. Res. Commun., 323, 668–673 (2004).
- 8) Hung L. M., Chen J. K., Huang S. S., Lee R. S., Su M. J., Cardiovasc. Res., 47, 549–555 (2000).
- 9) Valdameri G., Pereira Rangel L., Spatafora C., Guitton J., Gauthier C., Arnaud O., Ferreira-Pereira A., Falson P., Winnischofer S. M. B., Rocha M. E. M., Tringali C., Di Pietro A., Chem. Biol., 7, 322–330 (2000).
- 10) Murias M., Handler N., Erker T., Pleban K., Ecker G., Saiko P., Szekeres T., Jäger W., Bioorg. Med. Chem., 12, 5571–5578 (2004).
- 11) Dyer C., BMJ, 314, 993 (1997).
- 12) Ren H.-Y., Zhang X.-J., Song Z.-y., Rupert W., Gao G.-J., Guo S.-, Zhao L.-P., PLoS ONE, 6, e23258 (2011).
- 13) Grech J. N., Li Q., Roufogalis B. D., Duke C. C., J. Nat. Prod., 57, 1682–1687 (1994).
- 14) Chang M.-J., Xiao J.-H., Wang Y., Yan Y.-L., Yang J., Wang J.-L., PLoS ONE, 7, e50291 (2012).
- 15) Jiang Z., Xu J., Long M., Tu Z., Yang G., He G., Life Sci., 85, 345–350 (2009).
- 16) Ryu S., Han Y., Han B., Arch. Pharm. Res., 11, 230–239 (1988).
- 17) Wang X., Zhao L., Han T., Chen S., Wang J., Eur. J. Pharm., 578, 339–348 (2008).
- 18) Han X., Ling S., Gan W., Sun L., Duan J., Xu J.-W., Atherosclerosis, 225, 76–82 (2012).
- 19) Ren X.-l, Wang G.-f, Wang M., Ou-Yang H.-z, Qi A.-d, J. Pharm. Biomed. Anal., 55, 211–215 (2011).
- 20) Zhao Y.-Y., Cheng X.-L., Wei F., Han X.-Q., Xiao X.-Y., Lin R.-C., J. Liquid Chromatogr. Relat. Technol., 36, 717–730 (2012).
- 21) Regev-Shoshani G., Shoseyov O., Bilkis I., Kerem Z., Biochem. J., 374, 157–163 (2003).
- 22) Su D., Cheng Y., Liu M., Liu D., Cui H., Zhang B., Zhou S., Yang T., Mei Q., PLoS ONE, 8, e54505 (2013).
- 23) Ryu G., Ju J., Park Y., Ryu S., Choi B., Lee B., Arch. Pharm. Res., 25, 636–639 (2002).
- 24) Han Y., Ryu S., Han B., Arch. Pharm. Res., 13, 132–135 (1990).
- 25) Gangjee A., Namjoshi O. A., Keller S. N., Smith C. D., Bioorg. Med. Chem., 19, 4355–4365 (2011).
- 26) Caldwell S. T., McPhail D. B., Duthie G. G., Hartley R. C., Can. J. Chem., 90, 23–33 (2012).
- 27) Alonso F., Riente P., Yus M., Eur. J. Org. Chem., 2009, 6034–6042 (2009).
- 28) Ali M. A., Tsuda Y., Chem. Pharm. Bull., 40, 2842–2844 (1992).
- 29) Feutrill G. I., Mirrington R. N., Aust. J. Chem., 25, 1719–1729 (1972).
- 30) Farina A., Ferranti C., Marra C., Guiso M., Norcia G., Nat. Prod. Res., 21, 564–573 (2007).
- 31) Chalal M., Vervandier-Fasseur D., Meunier P., Cattey H., Hierso J.-C., Tetrahedron, 68, 3899–3907 (2012).
- 32) Typical procedure for the Mizoroki–Heck reaction of 10 and 11: To a stirred solution of 10 (116 mg, 0.53 mmol) in dry DMF (1.5 mL) kept at r.t. under argon were added dry Na2CO3 (169 mg, 1.60 mmol), tetrabutylammonium iodide (233 mg, 0.63 mmol), Pd(OAc)2 (5.9 mg, 5 mol %) and 11 (172 mg, 1.06 mmol). The mixture was heated to 100°C for 10 h. Then EtOAc (20 mL), benzene (10 mL) and H2O (20 mL) were added. The organic layer was separated and washed with H2O (20 mL) and dried over Na2SO4. After filtration and evaporation, the residue was chromatographed on silica gel (hexane/EtOAc 2 : 1) to provide the 2,3-dihydrobenzofuran 13 as a brown solid. mp: 80–83°C; TLC Rf=0.48 (1 : 1 hexane/EtOAc); IR (film) 3020, 1726, 1042 cm−1; 1H-NMR (400 MHz, CDCl3) δ 2.23 (s, 3H, COCH3), 3.11–3.18 (dd, J=9.0, 15.8 Hz, 1H, C3-H), 3.43–3.49 (dd, J=9.2, 15.6 Hz, 1H, C3-H), 3.77 (s, 3H, OCH3), 5.45 (br s, 1H, OH), 5.62–5.67 (t, J=9.0 Hz, 1H, C2-H), 6.43 (s, 1H, ArH), 6.44 (s, 1H, ArH), 6.66–6.69 (d, J=11.2 Hz, 2H, ArH), 7.16–7.19 (d, 2H, ArH), 13C-NMR (100 MHz, CDCl3) δ 21.1 (CH3), 38.6 (CH3), 56.0 (CH2), 85.2 (CH), 105.3 (CH), 110.0 (CH), 115.3 (CH), 127.8 (CH), 127.8 (CH), 133.0 (C), 144.0 (C), 144.5 (C), 145.6 (C), 155.7 (C), 170.5 (C), HRMS (Fab, 3-NBA) m/z Calcd for C17H17O5 (M+H+) 301.1076, Found 301.1110.
- 33) Schädel U., Habicher W. D., Heterocycles, 57, 1049–1055 (2002).
- 34) Larock R. C., Yang H., Pace P., Narayanan K., Russell C. E., Cacchi S., Fabrizi G., Tetrahedron, 54, 7343–7356 (1998).


