Experimental
1H- and 13C-NMR spectra were recorded on a JEOL JNM-ECA600 or JEOL JNM-ECA500, and the chemical shifts were expressed in δ (ppm) values with trimethylsilane as an internal reference (s=singlet, d=doublet, t=triplet, q=quartet, m=multiplet, and br=broad peak). Some NMR signals doubled and broadened due to the existence of rotamers caused by the sterically hindered amide moiety. MS were recorded on a micromass Platform LC or Shimadzu LCMS-2010EV. High resolution (HR) mass spectral data were acquired using a Shimadzu LCMS-IT-TOF equipped with an electrospray ionization (ESI)/atmospheric pressure chemical ionization dual ion source. The purities of the final compounds were confirmed using LC-MS on an Agilent instrument with ESI. The LC-MS conditions were as follows: Agilent 1290 infinity and Agilent 6150; column Waters Acquity CSH C18, 1.7 µm, 2.1 mm ×50 mm; eluent A, water +0.1% formic acid; eluent B, acetonitrile +0.1% formic acid; 20–99% B for 1.2 min, 99% B for 0.2 min; flow rate 0.8 mL/min; UV detection, λ=254 nm.
1-Methyl-N-{[3-(trifluoromethoxy)phenyl]methyl}-1H-imidazole-4-carboxamide (9)To a solution of 1-methyl-1H-imidazole-4-carboxylic acid (0.50 g, 4.0 mmol), 1-hydroxybenztriazole monohydrate (0.74 g, 4.8 mmol), and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (0.92 g, 4.8 mmol) in acetonitrile (10 mL) was added a solution of 3-(trifluoromethoxy)benzylamine 8 (0.84 g, 4.4 mmol) in acetonitrile (10 mL), and the mixture was stirred at room temperature for overnight. The reaction mixture was concentrated in vacuo, and saturated aqueous NaHCO3 solution was added to the residue. After extraction with ethyl acetate, the organic layer was dried over anhydrous MgSO4, filtered, and concentrated in vacuo. The residue was purified using silica gel column chromatography (0–10% MeOH in CHCl3) to obtain 9 (1.18 g, 98%) as a colorless powder. 1H-NMR (600 MHz, CDCl3) δ: 3.73 (3 H, s), 4.62 (2 H, d, J=6.0 Hz), 7.08–7.13 (1 H, m), 7.18 (1 H, s), 7.26–7.29 (1 H, m), 7.31–7.37 (2 H, m), 7.44–7.51 (1 H, m), 7.55 (1 H, d, J=1.4 Hz); MS ESI: m/z 300 [M+H]+.
N,1-Dimethyl-N-{[3-(trifluoromethoxy)phenyl]methyl}-1H-imidazole-4-carboxamide (6a)To a solution of 9 (50 mg, 0.17 mmol) in N,N-dimethylformamide (1 mL) was added sodium hydride (60% in oil, 8 mg, 0.20 mmol), and the mixture was stirred at room temperature for 10 min. To the reaction mixture was added methyl iodide (12 µL, 0.19 mmol), and the mixture was stirred for an additional 30 min. The reaction was quenched with saturated aqueous NaHCO3 solution and extracted with ethyl acetate. The organic layer was washed with water and brine, dried over anhydrous MgSO4, filtered, and concentrated in vacuo. The residue was purified using silica gel column chromatography (0–2% MeOH in CHCl3) to obtain 6a (42 mg, 80%) as a colorless powder. 1H-NMR (600 MHz, DMSO-d6) δ: 2.83 and 3.38 (3H, br s), 3.69 (3H, s), 4.67 and 5.37 (2H, br s), 7.23–7.32 (3H, m), 7.48 (1H, t, J=7.8 Hz), 7.64 (1H, br s), 7.69–7.72 (1H, m); 13C-NMR (151 MHz, DMSO-d6) δ: 33.12, 33.46, 36.23, 50.32, 52.21, 119.38, 119.64, 119.82, 120.03 (q, J=256.7 Hz), 126.53, 130.37, 136.84, 137.42, 140.96, 141.54, 148.52, 163.37; HR-MS: Calcd for C14H14F3N3O2 [M+H]+ 314.1111. Found 314.1089.
N-Ethyl-1-methyl-N-{[3-(trifluoromethoxy)phenyl]methyl}-1H-imidazole-4-carboxamide (6b)Compound 6b (73%, colorless powder) was obtained in a similar manner to that described for 6a. 1H-NMR (600 MHz, DMSO-d6) δ: 0.99–1.18 (3H, m), 3.29 and 3.92 (2H, br s), 3.69 (3H, s), 4.67 and 5.37 (2H, br s), 7.23–7.31 (2H, m), 7.33 (1H, d, J=7.8 Hz), 7.47 (1H, t, J=7.9 Hz), 7.61–7.69 (1H, m), 7.71 (1H, s); 13C-NMR (151 MHz, DMSO-d6) δ: 12.32, 14.39, 33.10, 40.45, 42.54, 47.81, 49.68, 119.26, 119.78, 120.04 (q, J=259.72 Hz), 126.43, 130.21, 137.06, 137.51, 141.79, 142.05, 148.46, 162.73, 163.27; HR-MS: Calcd for C15H16F3N3O2 [M+H]+ 328.1267. Found 328.1261.
1-Methyl-N-propyl-N-{[3-(trifluoromethoxy)phenyl]methyl}-1H-imidazole-4-carboxamide (6c)Compound 6c (73%, colorless oil) was obtained in a similar manner to that described for 6a. 1H-NMR (600 MHz, DMSO-d6) δ: 0.73–0.84 (3H, m), 1.45–1.62 (2H, m), 3.22 and 3.87 (2H, br s), 3.70 (3H, s) 4.68 and 5.39 (2H, br s), 7.23–7.34 (3H, m), 7.46 (1H, t, J=8.0 Hz), 7.59–7.68 (1H, m), 7.70 (1H, s); 13C-NMR (151 MHz, DMSO-d6) δ: 10.75, 11.13, 20.04, 21.86, 33.10, 47.31, 48.31, 49.28, 50.16, 119.26, 119.74, 120.04 (q, J=255.19 Hz), 126.39, 126.63, 127.20, 130.21, 137.04, 137.44, 139.35, 141.69, 142.05, 148.46, 163.39; HR-MS: Calcd for C16H18F3N3O2 [M+H]+ 342.1424. Found 342.1404.
1-Methyl-N-(2-methylpropyl)-N-{[3-(trifluoromethoxy)phenyl]methyl}-1H-imidazole-4-carboxamide (6e)Compound 6e (40%, colorless oil) was obtained in a similar manner to that described for 6a. 1H-NMR (499 MHz, CDCl3) δ: 0.82–0.98 (6H, m), 1.94–2.13 (1H, m), 3.21–3.28 and 3.88–3.98 (2H, m), 3.71 (3H, br s), 4.76 and 5.43 (2H, br s), 7.05–7.24 (3H, m), 7.29–7.35 (1H, m), 7.35–7.43 (1H, m), 7.57 (1H, d, J=1.4 Hz); 13C-NMR (126 MHz, CDCl3) δ: 19.90, 20.27, 26.71, 33.68, 51.58, 53.13, 119.41, 120.01, 126.37, 129.80, 136.47, 149.46, 164.04; HR-MS: Calcd for C17H20F3N3O2 [M+H]+ 356.1580. Found 356.1551.
N-{[3-(Trifluoromethoxy)phenyl]methyl}propan-2-amine (10a)To a solution of acetone (0.38 mL, 5.2 mmol) and 8 (1.0 g, 5.2 mmol) in CHCl3 (10 mL) was added sodium triacetoxyborohydride (2.2 g, 10 mmol), and the mixture was stirred at room temperature for 45 min. The mixture was quenched with saturated aqueous NaHCO3 solution and extracted with CHCl3. The organic layer was washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The residue was purified with silica gel column chromatography (0–10% MeOH in CHCl3) to obtain 10a (0.68 g, 56%) as a colorless oil. 1H-NMR (600 MHz, CDCl3) δ: 1.10 (6H, d, J=6.4 Hz), 2.84 (1H, spt, J=6.3 Hz), 3.80 (2H, s), 7.09 (1H, d, J=7.3 Hz), 7.20 (1H, s), 7.30–7.36 (1H, m); MS (ESI): m/z 234 [M+H]+.
N-{[3-(Trifluoromethoxy)phenyl]methyl}cyclohexanamine (10b)Compound 10b (quant., colorless oil) was obtained in a similar manner to that described for 10a. 1H-NMR (600 MHz, CDCl3) δ: 1.08–1.27 (4H, m), 1.56–1.66 (2H, m), 1.67–1.80 (2H, m), 1.82–1.99 (2H, m), 2.43–2.51 (1H, m), 3.83 (2H, s), 7.07–7.12 (1H, m), 7.20–7.28 (2H, m), 7.30–7.38 (1H, m); MS (ESI): m/z 274 [M+H]+.
N-{[3-(Trifluoromethoxy)phenyl]methyl}oxan-4-amine (10c)Compound 10c (82%, colorless oil) was obtained in a similar manner to that described for 10a. 1H-NMR (600 MHz, CDCl3) δ: 1.40–1.49 (2H, m) 1.82–1.89 (2H, m) 2.66–2.76 (1H, m) 3.36–3.44 (2H, m) 3.85 (2H, s) 3.94–4.02 (2H, m) 7.08–7.38 (4H, m); MS (ESI): m/z 276 [M+H]+.
1-Methyl-N-(propan-2-yl)-N-{[3-(trifluoromethoxy)phenyl]methyl}-1H-imidazole-4-carboxamide (6d)Compound 6d (93%, colorless powder) was obtained from 10a in a similar manner to that described for 9. 1H-NMR (600 MHz, DMSO-d6) δ: 1.02–1.15 (6H, m), 3.69 (3H, br s), 4.44–4.71 and 5.21–5.40 and 5.54–5.67 (3H, m), 7.18 (1H, br d, J=8.3 Hz), 7.22 (1H, br s), 7.31 (1H, d, J=7.8 Hz), 7.42 (1H, s), 7.54–7.72 (2H, m); 13C-NMR (151 MHz, DMSO-d6) δ: 19.89, 21.16, 33.08, 42.91, 46.68, 48.17, 118.65, 119.20, 120.02 (q, J=258.21), 125.79, 125.93, 126.51, 126.69, 129.85, 137.34, 143.39, 148.28, 156.52, 163.85; HR-MS: Calcd for C16H18F3N3O2 [M+H]+ 342.1424. Found 342.1398.
N-Cyclohexyl-1-methyl-N-{[3-(trifluoromethoxy)phenyl]methyl}-1H-imidazole-4-carboxamide (6f)Compound 6f (9%, colorless powder) was obtained from 10b in a similar manner to that described for 9. 1H-NMR (600 MHz, DMSO-d6) δ: 0.96–1.09 (1H, m), 1.13–1.59 (6H, m), 1.61–1.73 (3H, m), 3.70 (3H, br s), 4.12–4.32 and 4.54–4.76 and 5.06–5.46 (3H, m), 7.17 (1H, d, J=8.3 Hz), 7.20 (1H, s), 7.29 (1H, br d, J=7.4 Hz), 7.38–7.44 (1H, m), 7.48–7.72 (2H, m); 13C-NMR (151 MHz, DMSO-d6) δ: 24.72, 25.44, 30.02, 31.41, 33.08, 43.95, 46.79, 54.78, 56.31, 118.57, 119.12, 119.94, 120.01 (q, J=256.7 Hz), 125.75, 126.03, 126.63, 129.79, 130.15, 137.34, 137.67, 143.41, 144.04, 148.24, 148.42, 163.27, 164.05; HR-MS: Calcd for C19H22F3N3O2 [M+H]+ 382.1737. Found 382.1716.
1-Methyl-N-(oxan-4-yl)-N-{[3-(trifluoromethoxy)phenyl]methyl}-1H-imidazole-4-carboxamide (6g)Compound 6g (14%, colorless powder) was obtained from 10c in a similar manner to that described for 9. 1H-NMR (600 MHz, CDCl3) δ: 1.56–1.82 (4H, m), 3.45 (2H, br t, J=11.8 Hz), 3.71 (3H, br s), 3.95 (2H, br dd, J=11.6, 4.1 Hz), 4.60–4.86 and 5.28–5.66 (3H, m), 7.05 (1H, br d, J=7.8 Hz), 7.12 (1H, br s), 7.18–7.23 (1H, m), 7.26–7.46 (1H, m), 7.36–7.40 (1H, m), 7.57 (1H, br s); 13C-NMR (151 MHz, DMSO-d6) δ: 30.62, 32.06, 33.57, 44.98, 47.46, 52.54, 54.19, 67.47, 119.00, 119.53, 120.43 (q, J=259.7 Hz), 125.13, 126.48, 129.55, 136.63, 138.40, 142.18, 142.94, 149.31, 164.58; HR-MS: Calcd for C18H20F3N3O3 [M+H]+ 384.1530. Found 384.1507.
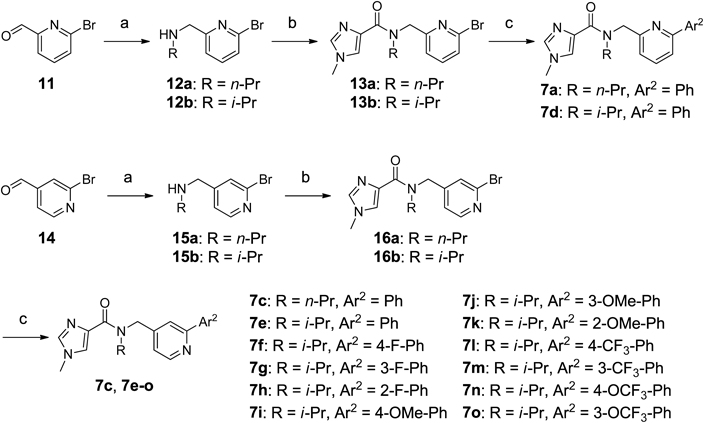
N-[(6-Bromopyridin-2-yl)methyl]propan-1-amine (12a)Compound 12a (99%, yellow oil) was obtained from 2-bromopyridine-6-carboxaldehyde 11 and 1-aminopropane in a similar manner to that described for 10a. 1H-NMR (600 MHz, CDCl3) δ: 0.94 (3H, t, J=7.3 Hz), 1.52–1.59 (2H, m), 2.59–2.64 (2H, m), 3.89 (2H, s), 7.29 (1H, d, J=7.4 Hz), 7.36 (1H, d, J=7.8 Hz), 7.51 (1H, t, J=7.5 Hz); MS (ESI): m/z 229 [M+H]+.
N-[(6-Bromopyridin-2-yl)methyl]propan-2-amine (12b)Compound 12b (49%, pale yellow oil) was obtained from 11 and 2-aminopropane in a similar manner to that described for 10a. 1H-NMR (600 MHz, CDCl3) δ: 1.11 (6H, d, J=6.0 Hz), 2.84 (1H, spt, J=6.3 Hz), 3.88 (2H, s), 7.29 (1H, d, J=7.3 Hz), 7.33–7.36 (1H, m), 7.49 (1H, t, J=7.6 Hz); MS (ESI): m/z 229 [M+H]+.
N-[(6-Bromopyridin-2-yl)methyl]-1-methyl-N-propyl-1H-imidazole-4-carboxamide (13a)Compound 13a (67%, pale yellow oil) was obtained from 12a in a similar manner to that described for 9. 1H-NMR (600 MHz, CDCl3) δ: 0.83–0.93 (3H, m), 1.61–1.71 (2H, m), 3.34–4.05 (5H, m), 4.72–5.51 (2H, m), 7.29–8.11 (5H, m); MS (ESI): m/z 337 [M+H]+.
N-[(6-Bromopyridin-2-yl)methyl]-1-methyl-N-(propan-2-yl)-1H-imidazole-4-carboxamide (13b)Compound 13b (95%, colorless powder) was obtained from 12b in a similar manner to that described for 9. 1H-NMR (600 MHz, CDCl3) δ: 1.09–1.24 (6H, m), 3.64–3.76 (3H, m), 4.70–4.88 and 5.29–5.36 and 5.68–5.75 (3H, m), 7.26–7.34 (2H, m), 7.41–7.47 (2H, m), 7.51–7.59 (1H, m); MS (ESI): m/z 337 [M+H]+.
N-[(2-Bromopyridin-4-yl)methyl]propan-1-amine (15a)Compound 15a (37%, pale yellow oil) was obtained from 2-bromopyridine-4-carboxaldehyde 14 and 1-aminopropane in a similar manner to that described for 10a. 1H-NMR (600 MHz, CDCl3) δ: 0.94 (1H, t, J=7.3 Hz), 1.50–1.55 (2H, m), 2.57 (2H, t, J=7.3 Hz), 3.79 (2H, s), 7.23 (1H, dd, J=5.0, 1.4 Hz), 7.49–7.50 (1H, m), 8.29 (1H, d, J=5.0 Hz); MS (ESI): m/z 229 [M+H]+.
N-[(2-Bromopyridin-4-yl)methyl]propan-2-amine (15b)Compound 15b (74%, pale yellow oil) was obtained from 14 and 2-aminopropane in a similar manner to that described for 10a. 1H-NMR (600 MHz, CDCl3) δ: 1.09 (6 H, d, J=6.4 Hz), 2.83 (1 H, spt, J=6.3 Hz), 3.78 (2 H, s), 7.23 (1 H, dd, J=5.0, 0.9 Hz), 7.50–7.51 (1 H, m), 8.28 (1 H, d, J=5.0 Hz); MS (ESI): m/z 229 [M+H]+.
N-[(2-Bromopyridin-4-yl)methyl]-1-methyl-N-propyl-1H-imidazole-4-carboxamide (16a)Compound 16a (81%, pale yellow oil) was obtained from 15a in a similar manner to that described for 9. 1H-NMR (600 MHz, CDCl3) δ: 0.84–0.97 (3H, m), 1.59–1.73 (2H, m), 3.29–4.04 (5H, m), 4.60–4.73 (1H, m), 5.30–5.50 (1H, m), 7.12–7.64 (4H, m), 8.28 (1H, d, J=5.0 Hz); MS (ESI): m/z 337 [M+H]+.
N-[(2-Bromopyridin-4-yl)methyl]-1-methyl-N-(propan-2-yl)-1H-imidazole-4-carboxamide (16b)Compound 16b (98%, pale yellow oil) was obtained from 15b in a similar manner to that described for 9. 1H-NMR (600 MHz, CDCl3) δ: 1.14–1.23 (6H, m), 3.62–3.77 (3H, m), 4.49–4.90 and 5.22–5.34 and 5.75–5.84 (3H, m), 7.17 (1H, br s), 7.27–7.31 (1H, m), 7.37–7.45 (2H, m), 7.57 (1H, br s), 8.24 (1H, br d, J=5.0 Hz); MS (ESI): m/z 337 [M+H]+.
1-Methyl-N-[(6-phenylpyridin-2-yl)methyl]-N-propyl-1H-imidazole-4-carboxamide (7a)To a solution of 13a (150 mg, 0.44 mmol) in N,N-dimethylformamide (2 mL) and ethanol (1 mL) were added phenylboronic acid (107 mg, 0.88 mmol), potassium carbonate (122 mg, 0.88 mmol), and tetrakis(triphenylphosphine)palladium(0) (51 mg, 0.044 mmol), and the reaction mixture was stirred at 150°C under microwave irradiation for 10 min. Water was added, and the mixture was extracted with ethyl acetate. The organic layer was washed with water and brine, dried over anhydrous MgSO4, filtered, and concentrated in vacuo. The residue was purified using silica gel column chromatography (0–5% MeOH in CHCl3) and NH-silica gel column chromatography (67–100% ethyl acetate in hexane) to obtain 7a (98 mg, 67%) as a colorless oil. 1H-NMR (499 MHz, CDCl3) δ: 0.84–0.95 (3H, m), 1.64–1.82 (2H, m), 3.46 (1H, br t, J=7.4 Hz), 3.68 and 3.72 (3H, br s), 4.07 (1H, br t, J=7.2 Hz), 4.93 (1H, br s), 5.53 (1H, s), 7.23–7.33 (2H, m), 7.38–7.49 (3H, m), 7.56–7.61 (2H, m), 7.68 (1H, t, J=7.7 Hz), 7.98–8.03 (2H, m); 13C-NMR (126 MHz, CDCl3) δ: 11.04, 11.45, 20.49, 22.24, 33.54, 48.56, 50.40, 52.01, 53.99, 118.65, 118.72, 119.71, 120.09, 125.90, 126.05, 126.95, 128.67, 128.77, 128.87, 136.58, 137.27, 138.61, 139.33, 139.51, 156.69, 158.14, 158.84, 164.20; HR-MS: Calcd for C20H22N4O [M+H]+ 335.1866. Found 335.1855.
1-Methyl-N-[(2-phenylpyridin-4-yl)methyl]-N-propyl-1H-imidazole-4-carboxamide (7c)Compound 7c (74%, colorless oil) was obtained from 16a in a similar manner to that described for 7a. 1H-NMR (499 MHz, CDCl3) δ: 0.83–0.98 (3H, m), 1.63–1.76 (2H, m), 3.41 (1H, br s), 3.69 and 3.72 (3H, br s), 3.99 (1H, br s), 4.79 (1H, br s), 5.49 (1H, br s), 7.09–7.20 (1H, m), 7.28–7.48 (4H, m), 7.57–7.66 (2H, m), 7.95 (2H, br d, J=6.9 Hz), 8.61 (1H, d, J=4.8 Hz); 13C-NMR (126 MHz, CDCl3) δ: 11.01, 11.42, 20.54, 22.28, 33.59, 48.49, 49.06, 50.13, 51.06, 119.06, 119.41, 120.76, 120.98, 126.50, 127.01, 128.66, 128.91, 136.65, 138.32, 139.33, 148.15, 148.96, 149.74, 157.69, 163.87, 164.32; HR-MS: Calcd for C20H22N4O [M+H]+ 335.1866. Found 335.1856.
1-Methyl-N-[(6-phenylpyridin-2-yl)methyl]-N-(propan-2-yl)-1H-imidazole-4-carboxamide (7d)Compound 7d (52%, colorless oil) was obtained from 13b in a similar manner to that described for 7a. 1H-NMR (499 MHz, CDCl3) δ: 1.14–1.30 (6H, m), 3.65 and 3.73 (3H, br s), 4.86 (2H, br s), 5.43 and 5.72 (1H, br s), 7.28 (1H, br d, J=7.5 Hz), 7.37–7.49 (4H, m), 7.53–7.57 (2H, m), 7.62–7.67 (1H, m), 8.00 (2H, d, J=7.5 Hz); 13C-NMR (126 MHz, CDCl3) δ: 20.23, 21.50, 33.57, 46.88, 47.26, 48.97, 49.99, 118.37, 119.77, 120.03, 125.86, 126.98, 128.43, 128.53, 128.64, 129.47, 131.91, 131.93, 132.05, 132.13, 136.57, 136.83, 137.11, 156.41, 159.28, 164.48; HR-MS: Calcd for C20H22N4O [M+H]+ 335.1866. Found 335.1855.
1-Methyl-N-[(2-phenylpyridin-4-yl)methyl]-N-(propan-2-yl)-1H-imidazole-4-carboxamide (7e)Compound 7e (58%, colorless amorphous) was obtained from 16b in a similar manner to that described for 7a. 1H-NMR (499 MHz, CDCl3) δ: 1.14–1.30 (6H, m), 3.66 and 3.72 (3H, br s), 4.69 and 5.37 (2H, br s), 4.78–4.94 and 5.74–5.86 (1H, m), 7.14–7.25 (1H, m), 7.35–7.49 (4H, m), 7.56–7.59 (1H, m), 7.64 (1H, br s), 7.94 (2H, br d, J=7.5 Hz), 8.57 (1H, br d, J=4.5 Hz); 13C-NMR (126 MHz, CDCl3) δ: 20.37, 21.61, 33.60, 43.88, 46.98, 47.27, 48.85, 109.90, 118.88, 120.51, 126.36, 127.01, 128.64, 128.81, 136.64, 138.37, 139.53, 149.51, 149.74, 150.96, 157.43, 164.48; HR-MS: Calcd for C20H22N4O [M+H]+ 335.1866. Found 335.1851.
N-{[2-(4-Fluorophenyl)pyridin-4-yl]methyl}-1-methyl-N-(propan-2-yl)-1H-imidazole-4-carboxamide Bishydrochloride (7f)To a solution of 16b (1.50 g, 4.4 mmol) in N,N-dimethylformamide (10 mL) and ethanol (5 mL) were added 4-fluorophenylboronic acid (1.24 g, 8.9 mmol), potassium carbonate (1.23 g, 8.9 mmol), and tetrakis(triphenylphosphine)palladium(0) (0.51 g, 0.45 mmol), and the reaction mixture was stirred at 150°C under microwave irradiation for 30 min. Water was added, and the mixture was extracted with CHCl3. The organic layer was dried over anhydrous MgSO4, filtered, and concentrated in vacuo. The residue was purified using NH-silica gel column chromatography (25–100% ethyl acetate in hexane) and silica gel column chromatography (0–10% MeOH in CHCl3). The fractions including the product were collected and concentrated in vacuo. The residue was dissolved in ethyl acetate (10 mL), and 4 mol/L HCl in ethyl acetate (1.9 mL) was added to the solution. After stirring at room temperature for 4.5 h, the resultant precipitate was collected by filtration to obtain 7f (1.41 g, 79%) as a colorless powder. 1H-NMR (600 MHz, DMSO-d6) δ: 1.11–1.31 (6H, m), 3.75 and 3.92 (3H, br s), 4.61 and 4.89 and 5.12 (3H, br s), 7.46 (2H, t, J=8.7 Hz), 7.73 (1H, br s), 8.18–8.28 (4H, m), 8.72 (1H, d, J=5.8 Hz), 9.20 (1H, br s); 13C-NMR (151 MHz, DMSO-d6) δ: 20.90, 35.57, 44.09, 49.94, 116.14, 116.28, 121.37, 122.23, 124.02, 126.61, 129.61, 130.57, 137.10, 144.08, 151.33, 156.70, 159.43, 163.05, 164.70; HR-MS: Calcd for C20H21FN4O [M+H]+ 353.1772. Found 353.1784.
N-{[2-(3-Fluorophenyl)pyridin-4-yl]methyl}-1-methyl-N-(propan-2-yl)-1H-imidazole-4-carboxamide (7g)Compound 7g (49%, colorless amorphous) was obtained from 16b in a similar manner to that described for 7a. 1H-NMR (499 MHz, CDCl3) δ: 1.14–1.30 (6H, m), 3.68 and 3.73 (3H, br s), 4.61–4.92 and 5.24–5.46 and 5.81 (3H, m), 7.08 (1H, td, J=8.2, 2.4 Hz), 7.20 (1H, br s), 7.33–7.50 (2H, m), 7.55–7.65 (2H, m), 7.65–7.74 (2H, m), 8.57 (1H, br d, J=4.8 Hz); 13C-NMR (126 MHz, CDCl3) δ: 20.40, 21.61, 33.62, 43.85, 46.98, 47.32, 48.89, 113.86, 114.04, 115.55, 115.73, 118.94, 121.06, 122.54, 126.41, 128.44, 128.54, 130.06, 130.13, 136.67, 141.79, 149.57, 156.03, 162.27, 164.21; HR-MS: Calcd for C20H21FN4O [M+H]+ 353.1772. Found 353.1784.
N-{[2-(2-Fluorophenyl)pyridin-4-yl]methyl}-1-methyl-N-(propan-2-yl)-1H-imidazole-4-carboxamide Bishydrochloride (7h)To a solution of 16b (150 mg, 0.44 mmol) in toluene (1.5 mL), ethanol (1.5 mL), and water (1.0 mL) were added 2-fluorophenylboronic acid (93 mg, 0.66 mmol), cesium carbonate (217 mg, 0.67 mmol), and tetrakis(triphenylphosphine)palladium(0) (26 mg, 0.022 mmol), and the reaction mixture was stirred at 150°C under microwave irradiation for 30 min. Water was added, and the mixture was extracted with ethyl acetate. The organic layer was dried over anhydrous MgSO4, filtered, and concentrated in vacuo. The residue was purified using silica gel column chromatography (0–10% MeOH in CHCl3). The fractions including the product were collected and concentrated in vacuo. The residue was dissolved in MeOH (2 mL), and 2 mol/L HCl in MeOH (0.8 mL) was added to the solution. The solution was concentrated in vacuo to obtain 7h (165 mg, 87%) as a colorless amorphous. 1H-NMR (600 MHz, DMSO-d6) δ: 1.21–1.30 (6H, m), 3.92 (3H, br s), 4.54–4.69 and 4.87–5.19 (3H, m), 7.42–7.50 (2H, m), 7.63–7.70 (1H, m), 7.81–7.98 (2H, m), 8.05 (1H, br s), 8.22–8.37 (1H, m), 8.83 (1H, d, J=5.8 Hz), 9.06–9.35 (1H, m); 13C-NMR (151 MHz, DMSO-d6) δ: 20.88, 35.65, 44.11, 48.53, 50.00, 116.42, 116.58, 123.02, 123.96, 124.42, 125.11, 126.11, 128.72, 128.80, 131.50, 132.04, 133.16, 137.14, 144.40, 148.02, 158.53, 159.19, 160.20; HR-MS: Calcd for C20H21FN4O [M+H]+ 353.1772. Found 353.1784.
N-{[2-(4-Methoxyphenyl)pyridin-4-yl]methyl}-1-methyl-N-(propan-2-yl)-1H-imidazole-4-carboxamide Bishydrochloride (7i)Compound 7i (73%, colorless powder) was obtained from 16b in a similar manner to that described for 7h. 1H-NMR (600 MHz, DMSO-d6) δ: 1.07–1.24 (6H, m), 3.66–3.81 (3H, m), 3.83 (3H, s), 4.73 (2H, br s), 5.26 (1H, br s), 7.08–7.12 (2H, m), 7.34 (1H, br d, J=5.4 Hz), 7.74–7.90 (1H, m), 7.93 (1H, s), 8.03 (2H, br d, J=8.7 Hz), 8.27 (1H, br s), 8.57 (1H, d, J=5.4 Hz); 13C-NMR (151 MHz, DMSO-d6) δ: 19.79, 21.08, 34.08, 43.55, 48.88, 55.37, 114.43, 119.00, 120.62, 125.35, 128.20, 128.60, 137.38, 146.41, 153.72, 160.92, 162.10; HR-MS: Calcd for C21H24N4O2 [M+H]+ 365.1972. Found 365.1961.
N-{[2-(3-Methoxyphenyl)pyridin-4-yl]methyl}-1-methyl-N-(propan-2-yl)-1H-imidazole-4-carboxamide Bishydrochloride (7j)Compound 7j (59%, colorless powder) was obtained from 16b in a similar manner to that described for 7h. 1H-NMR (600 MHz, DMSO-d6) δ: 1.10–1.30 (6H, m), 3.66–3.93 (3H, m), 3.88 (3H, s), 4.64–5.21 (3H, m), 7.12–7.18 (1H, m), 7.51 (1H, t, J=7.8 Hz), 7.58–7.68 (2H, m), 7.70–7.75 (1H, m), 8.16 (2H, s), 8.70 (1H, d, J=5.8 Hz), 8.98 (1H, br s); 13C-NMR (151 MHz, DMSO-d6) δ: 19.69, 20.94, 35.19, 43.91, 49.64, 55.43, 112.73, 116.70, 119.82, 121.03, 122.01, 124.36, 126.33, 130.29, 135.42, 137.20, 144.92, 152.56, 159.73, 160.07; HR-MS: Calcd for C21H24N4O2 [M+H]+ 365.1972. Found 365.1959.
N-{[2-(2-Methoxyphenyl)pyridin-4-yl]methyl}-1-methyl-N-(propan-2-yl)-1H-imidazole-4-carboxamide Bishydrochloride (7k)Compound 7k (86%, colorless powder) was obtained from 16b in a similar manner to that described for 7h. 1H-NMR (600 MHz, DMSO-d6) δ: 1.15–1.30 (6H, m), 3.85 (3H, s), 3.92 (3H, br s), 4.64 and 4.96 (3H, br s), 7.20 (1H, t, J=7.4 Hz), 7.30 (1H, d, J=8.3 Hz), 7.61–7.73 (2H, m), 7.96 (1H, br s), 8.12 (1H, s), 8.26 (1H, br s), 8.83 (1H, d, J=5.8 Hz), 9.19 (1H, br s); 13C-NMR (151 MHz, DMSO-d6) δ: 20.84, 35.55, 44.29, 49.92, 56.01, 112.34, 119.86, 121.01, 122.97, 124.18, 125.25, 126.59, 131.28, 133.49, 137.18, 141.34, 148.94, 159.15, 159.45; HR-MS: Calcd for C21H24N4O2 [M+H]+ 365.1972. Found 365.1962.
1-Methyl-N-(propan-2-yl)-N-({2-[4-(trifluoromethyl)phenyl]pyridin-4-yl}methyl)-1H-imidazole-4-carboxamide Bishydrochloride (7l)Compound 7l (98%, colorless amorphous) was obtained from 16b in a similar manner to that described for 7f. 1H-NMR (600 MHz, DMSO-d6) δ: 1.15–1.30 (6H, m), 3.75 and 3.92 (3H, br s), 4.62 and 4.83 and 5.04 (3H, br s), 7.55 (1H, br s), 7.92 (2H, m, J=8.3 Hz), 8.11 (1H, s), 8.26 (1H, br s), 8.32 (2H, m, J=8.3 Hz), 8.72 (1H, d, J=5.4 Hz), 9.20 (1H, br s); 13C-NMR (151 MHz, DMSO-d6) δ: 20.96, 35.57, 43.81, 49.90, 120.32, 122.15, 123.92, 124.13 (q, J=247.8 Hz), 125.79, 126.67, 128.02, 129.73, 129.95, 137.10, 140.24, 147.49, 152.90, 159.29; HR-MS: Calcd for C21H21F3N4O [M+H]+ 403.1740. Found 403.1743.
1-Methyl-N-(propan-2-yl)-N-({2-[3-(trifluoromethyl)phenyl]pyridin-4-yl}methyl)-1H-imidazole-4-carboxamide Bishydrochloride (7m)Compound 7m (81%, colorless amorphous) was obtained from 16b in a similar manner to that described for 7h. 1H-NMR (600 MHz, DMSO-d6) δ: 1.17–1.30 (6H, m), 3.76 and 3.93 (3H, br s), 4.59 and 4.87 and 5.06 (3H, br s), 7.63 (1H, br s), 7.80–7.86 (1H, m), 7.92 (1H, br d, J=7.4 Hz), 8.23 (1H, s), 8.28 (1H, br s), 8.43 (1H, d, J=8.3 Hz), 8.47 (1H, s), 8.73 (1H, d, J=5.4 Hz), 9.26 (1H, br s); 13C-NMR (151 MHz, DMSO-d6) δ: 20.94, 35.63, 43.83, 49.98, 120.84, 122.24 (q, J=273.3 Hz), 122.25, 123.86, 124.00, 126.37, 126.75, 127.01, 129.76 (q, J=30.2 Hz), 130.21, 131.42, 136.32, 137.08, 146.43, 152.00, 153.94, 159.23; HR-MS: Calcd for C21H21F3N4O [M+H]+ 403.1740. Found 403.1742.
1-Methyl-N-(propan-2-yl)-N-({2-[4-(trifluoromethoxy)phenyl]pyridin-4-yl}methyl)-1H-imidazole-4-carboxamide Bishydrochloride (7n)Compound 7n (95%, colorless amorphous) was obtained from 16b in a similar manner to that described for 7f. 1H-NMR (600 MHz, DMSO-d6) δ: 1.14–1.30 (6H, m), 3.76 and 3.93 (3H, br s), 4.60 and 4.87 and 5.08 (3H, br s), 7.59 (2H, br d, J=8.3 Hz), 7.68 (1H, br s), 8.15 (1H, s), 8.23–8.29 (3H, m), 8.73 (1H, d, J=5.4 Hz), 9.23 (1H, br s); 13C-NMR (151 MHz, DMSO-d6) δ: 20.94, 35.61, 43.97, 49.94, 119.16, 120.88, 120.97, 121.37, 122.27, 123.94, 126.51, 129.91, 137.10, 145.46, 149.89, 151.79, 159.31; HR-MS: Calcd for C21H21F3N4O2 [M+H]+ 419.1689. Found 419.1690.
1-Methyl-N-(propan-2-yl)-N-({2-[3-(trifluoromethoxy)phenyl]pyridin-4-yl}methyl)-1H-imidazole-4-carboxamide Bishydrochloride (7o)Compound 7o (60%, colorless amorphous) was obtained from 16b in a similar manner to that described for 7h. 1H-NMR (600 MHz, DMSO-d6) δ: 1.15–1.30 (6H, m), 3.77 and 3.93 (3H, br s), 4.61 and 4.86 and 5.02 (3H, br s), 7.51–7.66 (2H, m), 7.73 (1H, t, J=8.1 Hz), 8.12 (1H, s), 8.15–8.18 (2H, m), 8.27 (1H, br s), 8.72 (1H, d, J=5.8 Hz), 9.25 (1H, br s); 13C-NMR (151 MHz, DMSO-d6) δ: 20.94, 35.63, 43.87, 49.96, 119.84, 120.10 (q, J=258.2 Hz), 120.72, 122.25, 122.61, 123.86, 126.49, 131.13, 137.10, 137.59, 146.43, 148.88, 151.88, 154.01, 159.19; HR-MS: Calcd for C21H21F3N4O2 [M+H]+ 419.1689. Found 419.1691.
4-Bromo-N-propylpyridine-2-carboxamide (18)Compound 18 (84%, colorless oil) was obtained from 4-bromopyridine-2-carboxylic acid 17 and 1-aminopropane in a similar manner to that described for 9. 1H-NMR (600 MHz, CDCl3) δ: 1.00 (3H, t, J=7.3 Hz), 1.62–1.69 (2H, m), 3.41–3.46 (2H, m), 7.57–7.60 (1H, m), 7.92–8.01 (1H, m), 8.35 (1H, d, J=5.0 Hz), 8.38 (1H, d, J=1.8 Hz); MS (ESI): m/z 243 [M+H]+.
4-Phenyl-N-propylpyridine-2-carboxamide (19)Compound 19 (88%, pale yellow oil) was obtained from 18 in a similar manner to that described for 7a. 1H-NMR (600 MHz, CDCl3) δ: 1.02 (3H, t, J=7.3 Hz), 1.65–1.72 (2H, m), 3.43–3.51 (2H, m), 7.28–8.60 (9H, m); MS (ESI): m/z 241 [M+H]+.
N-[(4-Phenylpyridin-2-yl)methyl]propan-1-amine (20)To a solution of 19 (0.35 g, 1.5 mmol) in tetrahydrofuran (10 mL) was added lithium aluminium hydride (0.28 g, 7.3 mmol), and the mixture was stirred at 70°C for 2 h. The reaction was quenched with 10% aqueous NaOH solution under ice cooling, dried over anhydrous MgSO4, filtered, and concentrated in vacuo. The residue was purified using NH-silica gel column chromatography (50–100% ethyl acetate in hexane) to obtain 20 (107 mg, 32%) as a yellow oil. 1H-NMR (600 MHz, CDCl3) δ: 0.95 (3H, t, J=7.6 Hz), 1.53–1.62 (2H, m), 2.65–2.69 (2H, m), 3.97 (2H, s), 7.38 (1H, dd, J=5.0, 1.8 Hz), 7.42–7.46 (1H, m), 7.46–7.51 (2H, m), 7.54 (1H, s), 7.63–7.67 (2H, m), 8.60 (1H, d, J=5.0 Hz); MS (ESI): m/z 227 [M+H]+.
1-Methyl-N-[(4-phenylpyridin-2-yl)methyl]-N-propyl-1H-imidazole-4-carboxamide (7b)Compound 7b (28%, colorless oil) was obtained in a similar manner to that described for 9. 1H-NMR (499 MHz, CDCl3) δ: 0.83–0.95 (3H, m), 1.64–1.75 (2H, m), 3.45 (1H, br t, J=7.4 Hz), 3.68 and 3.71 (3H, br s), 4.04 (1H, br t, J=6.9 Hz), 4.92 (1H, br s), 5.55 (1H, br s), 7.28–7.47 (5H, m), 7.54–7.63 (4H, m), 8.58 (1H, br d, J=4.8 Hz); 13C-NMR (126 MHz, CDCl3) δ: 11.01, 11.44, 20.45, 22.22, 33.55, 48.55, 50.26, 51.77, 53.85, 119.34, 119.92, 120.09, 120.22, 126.21, 127.07, 127.16, 129.00, 136.62, 138.31, 138.48, 149.05, 149.24, 149.36, 149.51, 158.79, 159.57, 164.01, 164.20; HR-MS: Calcd for C20H22N4O [M+H]+ 335.1866. Found 335.1858.
Calculation of CNS MPOThe CNS MPO score was calculated according to a previously reported method.19) The c Log P value was calculated by a software from Daylight Chemical Information Systems, Inc. The c Log D, pKa, and tPSA were calculated usign the software from ACD/Laboratories.
BiologyGlycine Uptake Inhibitory AssayGlioma T98G cells expressing human GlyT1 were used. The T98G cells were seeded at a density of 2.0×104 cells/well onto a 96-well plate and cultured overnight in a carbon dioxide incubator. The test compound was dissolved in 100% DMSO and then dissolved in 10 mM 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid (HEPES) buffer solution (pH 7.4) containing 150 mM sodium chloride, 1 mM calcium chloride, 5 mM potassium chloride, 1 mM magnesium chloride, 10 mM glucose, and 0.2% bovine serum albumin. After the medium for the cell culture was removed, the test compound and [3H]glycine (final concentration, 250 nM) were added to the cells and reacted at room temperature for 15 min. After the completion of the reaction, the labeled glycine solution was aspirated with a manifold. The cells were then lysed with 0.5 mol/L sodium hydroxide solution. The amount of intracellular glycine was determined by measuring the radio activity in the cell lysate using a liquid scintillation counter. The quantity of glycine uptake in the presence of 10 µM (N-[3-(4′-fluorophenyl)-3-(4′-phenylphenoxy)propyl])sarcosine (ALX5407) was defined as nonspecific uptake, and the specific uptake amount was determined by subtracting the nonspecific uptake amount from the total uptake amount in the absence of 10 µM ALX5407. The glycine uptake inhibitory activity (IC50 value) was calculated from an inhibition curve for test compound concentrations of 10−9 to 10−5.
Measurement of Glycine Concentration in CSF of RatsRats were anesthetized by isoflurane 1 h after oral administration of vehicle or 7n, and were sacrificed by cutting of an abdominal aorta. Then, CSF samples were collected from the cisterna magna using a 29-gauge needle. The CSF samples were centrifuged at 21500×g for 10 min at 4°C. The glycine standard and the CSF samples were derivatized with 1.33 mM of o-phthaldialdehyde at 10°C for 2.5 min, then injected into a high-performance liquid chromatography device equipped with an electrochemical detection system, ECD-300 (Eicom, Kyoto, Japan). Derivatized glycine was separated on a reversed phase Octa-Decyl-Silyl (ODS) column, Eicompak SC-5ODS (3.0 mm, i.d.×150 mm, Eicom, Kyoto, Japan), at 35°C with a flow rate of 0.5 mL/min. The electrode potential for the electrochemical detector was set at +600 mV against Ag/AgCl reference electrode. The mobile phase consisted of 0.1 M phosphate buffer solution (pH 6.0) containing 10.0% v/v acetonitrile and 5.0 mg/L ethylenediaminetetraacetic acid disodium salt.
All the studies were reviewed by the Taisho Pharmaceutical Co., Ltd., Animal Care Committee.