Results and Discussion
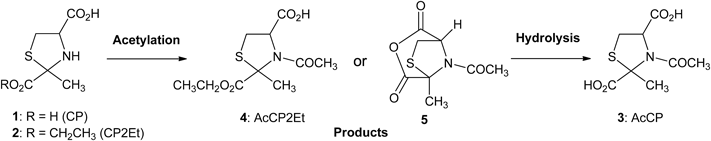
The synthesis of N-acetyl-2-methylthiazolidine-2,4-dicarboxylic acid (AcCP) derivatives is outlined in Chart 1. Derivatives of 2-methyl-tiazolidine-2,4-dicarboxylic acid (CP: 1a, b; CP2Et: 2a, b) were prepared by condensation of pyruvic acid or ethyl pyruvate with L-(R)-Cys. When these compounds are reacted in ethanol or water at room temperature (r.t.), a spontaneous condensation–cyclization reaction leads to the formation of a mixture of the two diastereoisomers of the CP derivatives (1a, b or 2a, b) in a ratio of ca. 1 : 1, as determined by 1H-NMR spectroscopy. These derivatives were converted to N-acetyl-2-methylthiazolidine-2,4-dicarboxylic acid derivatives (AcCP: 3, AcCP2Et: 4) under various reaction conditions as shown in Table 1. Since AcCP2Et (4) exists as four stereoisomers due to its two asymmetric centers at C2 and C4, characterization of each stereoisomer is complicated. Therefore, their stereochemistry was assigned on the basis of their analytical and spectral data. A reversed-phase HPLC (RP-HPLC) analysis and a chiral HPLC analysis were performed to evaluate the stereoisomer ratios of AcCP2Et (4a–d), and typical chromatograms are shown in Supplementary Fig. S1.
Table 1. Reaction of Thiazolidine Derivatives under Various Conditions
 |
|---|
| Entry | Substrate (ratio) | Acetylation | Products (ratio) | Yield (%) | Hydrolysis | 3: AcCP (ratio) | Yield (%) |
|---|
| 1 | 1a, b (1 : 1) | (CH3CO)2O/—a) | 5a, c (—) | 53.1 | HCl (aq)e) | 3a, c (1 : 1) | 21.7g) |
| 2 | 2a, b (1 : 1) | CH3COCl/Et3Nb) | 4b, d (1 : 1) | 58.8 | NaOH (aq)f) | 3b, d (1 : 1) | 66.8 |
| 3 | 2a, b (1 : 1) | (CH3CO)2O/—c) | 4b, d (1 : 1) | 46.5 | — | — | — |
| 4 | 2a, b (1 : 1) | CH3COCl/K2CO3d) | 4a, b (1 : 1) | 93.6 | NaOH (aq)f) | 3a, b, d (45 : 50 : 5) | 93.1g) |
| 5 | 2c, d (1 : 1) | CH3COCl/K2CO3d) | 4c, d (1 : 1) | 93.2 | — | — | — |
Reagents and conditions: a) (CH3CO)2O (4 eq.), 100°C, 2 h; b) CH3COCl (1.5 eq.), Et3N (2 eq.), AcOEt, rt, overnight; c) (CH3CO)2O (2 eq.), AcOEt, 90°C, 3 h; d) CH3COCl (1.2 eq.), K2CO3 (1.2 eq.), AcOEt, r.t., overnight; e) 5% citric acid, r.t., overnight, then added conc. HCl, 70°C, 1 h; f) 4 N NaOH (2.5 eq.), EtOH, 60°C, overnight; g) total yield.
Entry 1 in Table 1 shows the reaction of a ca. 1 : 1 mixture of CP (1a, b) and acetic anhydride. When CP (1a, b) and acetic anhydride was heated at 100°C according to Schubert’s method,25) a dark brown homogeneous solution formed in 2 h. The acid anhydride of AcCP (5) was found to be stable to silica gel column chromatography and isolated in modest yield. Acidic hydrolysis of 5 gave a single stereoisomer, i.e., the cis-isomer of AcCP (3), as indicated by 1H-NMR spectroscopy. In addition, this compound proved to be a mixture of cis-enantiomers of AcCP (3a, c) based on chiral HPLC analysis and the single crystal X-ray structure analysis described below (Fig. 1A). As reported by Szilágyi and Györgydeák,21) the N-acetylation of cis-(2R,4R)-CP (1a) proceeded with retention of configuration at C2 and C4 to give intramolecular acid anhydride 5a, whereas N-acetylation of trans-(2S,4R)-CP (1b) proceeded with epimerization at C4 via intermolecular mixed acid anhydride such as 6b and c followed by formation of intramolecular acid anhydride 5c as shown in (A) in Chart 2.
Acetylation of 2-Methylthiazolidine-2,4-dicarboxylic Acid 2-Ethyl Ester (CP2Et: 2a, b) with Acetyl Chloride and Organic AmineEntry 2 in Table 1 shows the reaction of a ca. 1 : 1 mixture of CP2Et (2a, b), derived from L-(R)-Cys and ethyl pyruvate, with acetyl chloride and Et3N. Contrary to CP (1a, b), CP2Et (2a, b) is highly soluble in ethyl acetate (AcOEt), and was acetylated by acetyl chloride in the presence of Et3N at r.t. When 1.5 equiv. of acetyl chloride and 2.0 equiv. of Et3N were used, acetylation completed to give AcCP2Et (4) as a single stereoisomer, as indicated by 1H-NMR spectroscopy, in satisfactory isolated yield. However, the existence of two stereoisomers was indicated by chiral HPLC. Similar results were obtained when other organic amines such as pyridine, N-methylmorpholine, or diisopropylethylamine were used instead of Et3N.
Then, AcCP2Et (4) was subjected to alkaline saponification at 60°C for 16 h to give a single stereoisomer of AcCP (3) by 1H-NMR, which was characteristically different from that of the above mentioned cis-enantiomers of AcCP (3a, c). The trans relative stereochemistry of the obtained AcCP (3) was estimated by comparing its 1H-NMR signals with those of cis-enantiomers of AcCP (3a, c). In addition, the single crystal X-ray structure analysis described below revealed that this compound is a mixture of enantiomers of trans-isomers, i.e., trans-(2S,4R)-AcCP (3b) and trans-(2R,4S)-AcCP (3d) (Fig. 1B). It is concluded that the trans-enantiomers of AcCP2Et (4b, d) were obtained under these acetylation conditions. Therefore, it is proposed that the configuration at C4 of cis-(2R,4R)-AcCP2Et (4a) is inverted to give the thermodynamically stable trans-(2R,4S)-AcCP2Et (4d) via an intermolecular mixed acid anhydrides (7a, d) (Chart 2B).
In addition, acetylation of CP2Et (2a, b) with more than 2 equiv. acetic anhydride without amine in refluxing ethyl acetate also gave an enantiomeric mixture of trans-enantiomers of AcCP2Et (4b, d) (Table 1, entry 3). The presence of their anhydrides with acetic acid such as 7d (Chart 2B) were confirmed by 1H-NMR spectroscopy of the crude reaction mixture. This observation strongly suggests the involvement of a mixed acid anhydride in the epimerization. Such a mechanism is consistent with that proposed in a study by Szilágyi and Györgydeák, in which selective epimerization at C4 of thiazolidine-4-carboxylic acids occurred via an intermediate formation of thiazolidine-4-carboxylic acid-acetic acid mixed anhydrides.21)
Acetylation of 2-Methylthiazolidine-2,4-dicarboxylic Acid 2-Ethyl Ester (CP2Et: 2a, b) with Acetyl Chloride and Inorganic BaseEntry 4 in Table 1 shows the reaction of a ca. 1 : 1 mixture of CP2Et (2a, b) with acetyl chloride in the presence of potassium carbonate (K2CO3) in ethyl acetate at r.t. Under these reaction conditions, more than 1.2 equiv. of acetyl chloride and 1.2 equiv. of K2CO3 were required to complete the reaction in satisfactory yield. Tetrahydrofuran (THF) or CH2Cl2 were found to be suitable solvents for this reaction, and potassium hydrogen carbonate, sodium carbonate, and sodium hydrogen carbonate were assessed as the inorganic base. Following these reaction conditions, a ca. 1 : 1 mixture of the cis- and trans-isomers of AcCP2Et (4) was indicated by 1H-NMR. Chiral HPLC analysis revealed that they were the cis-(2R,4R)-AcCP2Et (4a) and trans-(2S,4R)-AcCP2Et (4b). Therefore, it was concluded that the formation of a mixed acid anhydride and the epimerization at C4 are avoided under these conditions using an inorganic base as in the case of the 4-methyl ester derivative reported by Cremonesi et al.,24) and that N-acetylation proceeded with retention of the C4 configuration. Similarly, another set of stereoisomers of CP2Et (2c, d) obtained by the reaction of D-(S)-Cys and ethyl pyruvate gave a mixture of cis-(2S,4S)- and trans-(2R,4S)-AcCP2Et (4c, d) as shown in entry 5 in Table 1.
Hydrolysis of cis-(2R,4R)- and trans-(2S,4R)-AcCP2Et (4a, b) under alkaline conditions at 60°C for 16 h gave cis-(2R,4R)- and trans-(2S,4R)-AcCP (3a, b). In addition, formation of ca. 5% of the trans-(2R,4S)-AcCP (3d) was observed (Table 1, entry 4). Therefore, the C4 asymmetric center of cis-(2R,4R)-AcCP2Et (4a) and/or cis-(2R,4R)-AcCP (3a) are liable to isomerize under basic conditions to generate the stable trans-(2R,4S)-AcCP (3d).
Synthesis of Various Derivatives of 2-Methylthiazolidine-2,4-dicarboxylic AcidChart 3 shows examples of the derivatives of 2-methylthiazolidine-2,4-dicarboxylic acid produced using CP (1a, b), CP2Et (2a, b), and 2-methylthiazolidine-2,4-dicarboxylic acid 2-methyl ester (CP2Mt, 8). These derivatives were expected to possess different lipophilicities depending on the introduced functional groups. Reaction of CP2Et (2a, b) with a long-chain fatty acid chloride yielded surfactant-like prodrugs such as N-hexadecanoyl derivatives 11 and 12. The stereo-selectivity of N-acylation with a long-chain fatty acid chloride is similar to that of N-acetylation with acetyl chloride, and depends on the amount of acylation reagent and the kind of base used. Esterification of the 2-carboxylic acid group in cis-(2R,4R)- and trans-(2S,4R)-AcCP2Et (4a, b) with an alcohol afforded more lipophilic prodrug forms of Cys, such as 10a and b. Reaction of AcCP2Et (4a, b) with glycine (Gly) methyl ester or alanine (Ala) methyl ester afforded peptide-like derivatives, such as 13a and b. Of the synthesized compounds, AcCP (3), AcCP2Et (4), and AcCP2Me (9) show excellent water solubility and are suitable for assessing melanogenesis inhibition activity.

Chart 3. Synthesis of N-Acyl-2-methyl-2,4-thiazolidinedicarboxylic Acid Derivatives
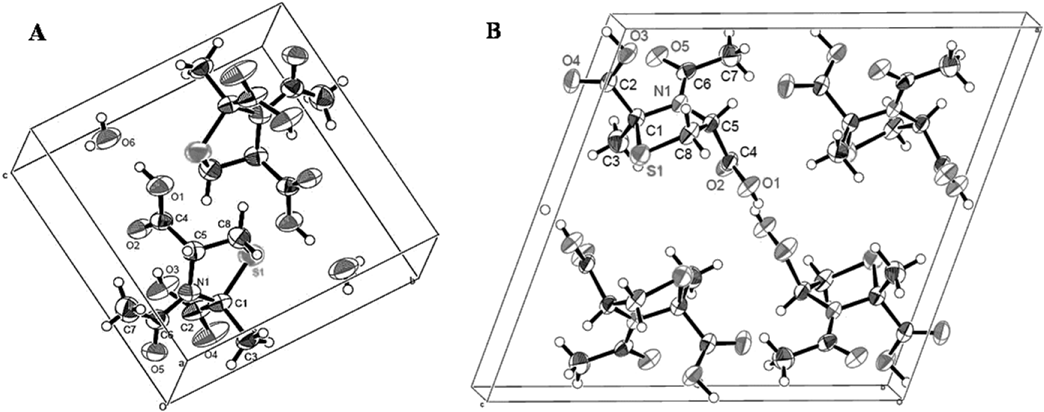
Crystal structures of the mixture of cis-enantiomers of AcCP (3a, c) (Table 1, entry 1) and the mixture of trans-enantiomers of AcCP (3b, d) (Table 1, entry 3) were determined by single crystal X-ray structure analysis. A summary of the crystallographic analyses is shown in Supplementary Table S1.
Figure 1A illustrates the crystal packing in the racemic crystal of cis-(2R,4R)- and cis-(2S,4S)-AcCP (3a, c) as an ORTEP drawing. This crystal belongs to the space group P-1 and its unit cell contains one molecule of cis-(2R,4R)-AcCP (3a), one molecule of cis-(2S,4S)-AcCP (3c), and two water molecules. The cis-enantiomers of AcCP (3a, c) molecule forms an intramolecular hydrogen bond between oxygen atoms O2 and O3, and adopts a relatively globular conformation. The hydrogen bonding network between a water molecule and oxygen atoms O1, O4, and O5 of neighboring cis-enantiomers of AcCP (3a, c) molecules plays an important role in the crystal packing.
Figure 1B demonstrates the crystal packing in the racemic crystal of trans-(2S,4R)- and trans-(2R,4S)-AcCP (3b, d) as an ORTEP drawing. This crystal belongs to the space group P21/c and its unit cell contains two molecules of trans-(2S,4R)-AcCP (3b) and two molecules of trans-(2R,4S)-AcCP (3d). Contrary to the cis-enantiomers of AcCP (3a, c) structure, the trans-enantiomers of AcCP (3b, d) molecule adopts a relatively extended conformation. Two kinds of intermolecular hydrogen bond, i.e., the hydrogen bond between O1 atom and O2 atom of neighboring enantiomer, and the hydrogen bond between O3 atom and O5 atom of neighboring enantiomer, play an important role in the crystal packing.
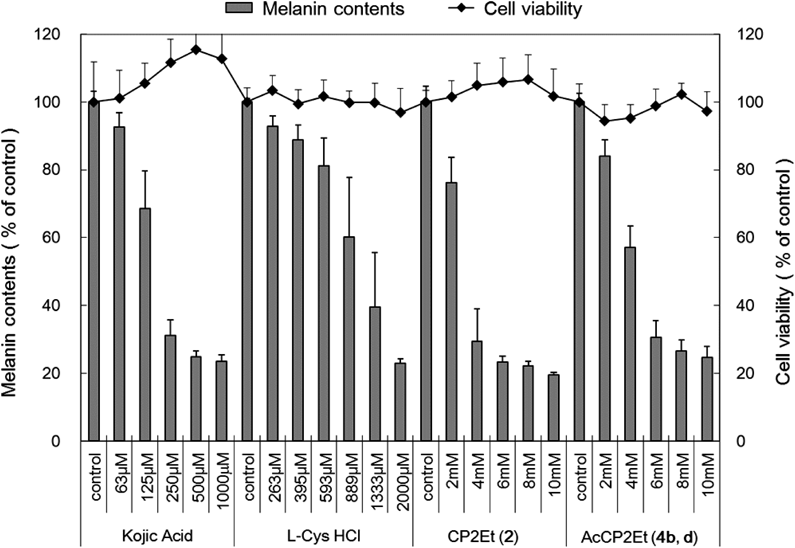
Suppression of Eumelanin ProductionFigure 2 shows the cell viabilities and the melanogenesis inhibition activity of the examined compounds.
We treated B16 melanoma cells with L-Cys, CP2Et (2a, b), and trans-enantiomers of AcCP2Et (4b, d) (Table 1, entry 2) to determine whether the compounds have cytotoxic effects. Cell viabilities were determined using Neutral Red (NR) assays. The results show that Cys is not cytotoxic at 2 mM, and that CP2Et (2a, b) and AcCP2Et (4b, d) are not cytotoxic at concentrations of 2, 4, 8, and 10 mM to B16 melanoma over 3 d.
B16 melanoma cells were incubated with various concentrations of kojic acid (a positive control), Cys, CP2Et (2a, b), and AcCP2Et (4b, d) under the same conditions for 3 d, and the melanin contents were measured to determine the melanogenesis inhibition of the test molecules. Cys, CP2Et (2a, b), and AcCP2Et (4b, d) were found to reduce the melanin content in a concentration-dependent manner with IC50 values of 1108, 3120, and 4544 µM, respectively. The activity of AcCP2Et (4b, d) is estimated to be approximately one fourth of that of Cys under these experimental conditions. As the stability of the compounds increases, their inhibition activity decreases. Cys is considered to be a key compound in the control of melanogenesis, as it regulates tyrosinase activity as well as the ability of L-dopaquinone to generate eumelanin or pheomelanine.7,14) We believe that the Cys liberated by degradation of CP2Et (2a, b) or AcCP2Et (4b, d) is responsible for the melanogenesis inhibition observed. In the case of CP2Et (2a, b), it is thought that Cys released by extracellular non-enzymatic degradation is taken up into cells. Conversely, it is expected that AcCP2Et (4b, d) is first taken up into the cells; Cys is then liberated inside the cells by enzymatic cleavage of its amide bond and/or ester bond and followed by non-enzymatic ring opening. Since AcCP2Et (4b, d) is more active than trans-enantiomers of AcCP (3b, d) (data not shown), AcCP2Et (4b, d) may be more easily taken up into cells, which may be responsible for its higher activity relative to AcCP (3b, d). These data suggest that AcCP2Et (4b, d) may have the potential as a stable derivative of Cys for skin whitening. Research on the detection of Cys released from AcCP2Et (4b, d) in a skin model incorporating melanocytes is currently under investigation in our group, and the results will be published in due course.
Stability of N-Acetyl-2-methylthiazolidine-2,4-dicarboxylic Acid 2-Ethyl Ester (AcCP2Et, 4a, c)As mentioned above, alkaline hydrolysis of the ethyl ester group at the quaternary C2 in AcCP2Et (4) is slow. Therefore, it was assumed that AcCP2Et (4) would be stable in neutral or weakly acidic conditions. The stability of a ca. 1 : 1 mixture (20 mg/20 mL) of the cis-(2R,4R)- and trans-(2S,4R)-isomers of AcCP2Et (4a, b) (Table 1, entry 4) over a pH range of 4 to 7 was investigated, and the results are shown in Table 2. No degradation was observed at 70°C after 5 d for the AcCP2Et (4b) at pH 5 to 7 or for the AcCP2Et (4a) at pH 7. Three to 7% degradation was observed for the AcCP2Et (4b) at pH 4 and for the AcCP2Et (4a) at pH 5 and 6. A considerable degree of hydrolysis of the ethyl ester functionality in the AcCP2Et (4a) was observed at pH 4. Therefore, the AcCP2Et (4b) is quite stable to prolonged storage at pH 5 to 7. It is noteworthy that the degradation products are cis-(2R,4R)- and trans-(2S,4R)-isomers of AcCP (3a, b), which is also expected to function as a prodrug of Cys. Stability to enzymatic hydrolysis is currently under investigation in our group, and the results will be reported in due course.
Table 2. Time–Course Stability Test of AcCP2Et (
4a,
b) at 70°C
| pH | Residual values (%) |
|---|
| 4a (cis-Isomer) | 4b (trans-Isomer) |
|---|
| 4 | 69.6 | 95.8 |
| 5 | 93.5 | 99.8 |
| 6 | 96.9 | >99.9 |
| 7 | >99.9 | >99.9 |
The residual values (%) after five days are shown. Sample concentration: 20 mg sample in 20 mL of 25 mM phosphate buffer.
Aqueous solutions (1%) at pH 4 and 7 of trans-enantiomers of AcCP2Et (4b, d) (Table 1, entry 2) and CP2Et (2a, b) in sealed vials were kept in a thermostatic bath at 70°C for 6 d. The odor of each sample was evaluated by six panelists, and the evaluation scores of the six panelists were totaled (see Experimental). CP2Et (2a, b) is in equilibrium with Cys and ethyl pyruvate in aqueous solution via non-enzymatic ring opening and hydrolysis; therefore odor originating for Cys may be detected. Indeed, a strong sulfur odor was detected in both samples of CP2Et (2a, b), while a slight sulfur odor was detected in AcCP2Et (4b, d) at pH 4 and almost no odor was detected at pH 7. It was concluded that AcCP2Et (4b, d) shows less sulfur odor due to decomposition, and exhibits superior preservation stability to CP2Et (2a, b) on storage. Therefore, AcCP2Et is a useful compound with potential as a cosmetic ingredient.
Experimental
1H-NMR spectra were recorded using a Brucker Avance 400 (400 MHz) and electron spray ionization (ESI)-MS spectra were measured using a Thermo Quest TSQ 700. The melting point (mp) of the obtained crystal was measured using an IA9100 digital melting point measuring apparatus manufactured by Electrothermal (U.K.).
RP-HPLC ConditionsColumn: Inertsil ODS-3 (high-pressure type, GL Sciences, Tokyo, Japan, particle size 3 µm, inner diameter 4.6 mm, length 250 mm). Detector: UV absorption spectrophotometer (measurement wavelength: 210 nm). Eluent: A: 0.05 M KH2PO4 (adjusted to pH 2 with 85% H3PO4), B: MeOH. Gradient conditions: 0–5 min: A 100%, 5–25 min: A : B=50 : 50, 25–35 min: A : B=50 : 50, 35–40 min: A 100%, 40–45 min: A 100%. Flow rate: 0.8 mL/min. Column temperature: 30°C. Sample concentration: 25–50 mg/dL. Injection volume: 20 µL. Retention times of the stereoisomers of N-AcCP2Et (4): 4a, c (cis-isomers): 30.5 min; 4b, d (trans-isomers): 32.4 min.
Chiral HPLC ConditionsColumn: CHIRALPACK QN-AX (0.46 cm×15 cm). Buffer: MeOH–AcOH–AcONH4=98 : 2 : 0.5 (v/v/w). Flow rate: 1.0 mL/min. Temperature: 40°C. Detection: UV 250 nm. Sample concentration: 1% in MeOH. Injection: 10 µL. Retention time of stereoisomers of N-AcCP2Et (4): 4a (cis-(2R,4R)): 3.62 min; 4c (cis-(2S,4S)): 3.75 min; 4b (trans-(2S,4R)): 4.08 min; 4d (trans-(2R,4S)): 4.42 min.
2-Methylthiazolidine-2,4-dicarboxylic Acid (CP, 1a, b)Under an argon atmosphere, L-Cys (15.0 g, 124 mmol) was dissolved in dry ethanol (35 mL), pyruvic acid (18.6 mL) was added at r.t., and the mixture was stirred for 3 h. The resulting precipitate was collected by filtration, washed with ice-cooled ethanol, and dried under reduced pressure to give 1a and b (ca. 1 : 1 diastereomer mixture, 23.0 g, 120 mmol, 97% yield).
1H-NMR (dimethyl sulfoxide (DMSO)-d6) δ: 1.59 (1.5H, s), 1.72 (1.5H, s), 2.76 (0.5H, dd, J=10.1, 10.2 Hz), 2.97 (0.5H, dd, J=8.4, 9.9 Hz), 3.26 (0.5H, dd, J=6.7, 10.0 Hz), 3.40 (0.5H, dd, J=6.0, 10.2 Hz), 3.98 (0.5H, dd, J=6.0, 10.1 Hz), 4.19 (0.5H, dd, J=6.7, 8.3 Hz). ESI-MS m/z: 190.0 (M−H)−.
2-Methylthiazolidine-2,4-dicarboxylic Acid 2-Ethyl Ester (CP2Et, 2a, b)Under an argon atmosphere, L-Cys hydrochloride (60.0 g, 381 mmol) was added to ion-exchanged water (500 mL), and the pH of the solution was adjusted to 5.05 by adding aqueous sodium hydroxide (4 N, ca. 88 mL). Ethyl pyruvate (63.0 mL, 567 mmol) was added to the solution and the mixture was stirred for 4 h at r.t. The mixture was concentrated under reduced pressure to ca. 150 mL. Water (100 mL) was added, and the mixture was extracted with ethyl acetate (500, 200 mL). The organic layer was washed with saturated sodium chloride solution (50 mL), dried over anhydrous MgSO4, and concentrated to ca. 200 mL. To the residue was added n-heptane (220 mL) at 45°C and the mixture was stored overnight at r.t. The precipitate was collected and dried under reduced pressure to give 2a and b (ca. 1 : 1 diastereomer mixture, 65.6 g, 299 mmol, 79% yield).
1H-NMR (DMSO-d6) δ: diastereomer 1; 1.23 (3H, t, J=7.1 Hz), 1.62 (3H, s), 2.81 (1H, dd, J=8.6, 10.0 Hz), 3.42 (1H, dd, J=6.1, 10.4 Hz), 4.03 (1H, dd, J=6.2, 10.0 Hz), 4.20 (2H, dd, J=7.1, 13.9 Hz). Diastereomer 2; 1.19 (3H, t, J=7.1 Hz), 1.75 (3H, s), 2.97 (1H, dd, J=8.6, 10.0 Hz), 3.29 (1H, m), 4.09 (2H, m), 4.16 (1H, m). ESI-MS m/z: 220.0 (M+H)+.
N-Acetyl-2-methylthiazolidine-2,4-dicarboxylic Acid (cis-Isomer of AcCP, 3a, c) (Table 1, Entry 1)Compounds 1a and b (20.0 g, 105 mmol) and acetic anhydride (40 mL, 418 mmol) were heated at 100°C for 2 h. The reaction mixture was concentrated under reduced pressure, aqueous citric acid (5%, 200 mL) was added thereto, and the mixture stirred overnight at r.t. To the mixture was added concentrated hydrochloric acid (2.0 mL) and the mixture was stirred at 70°C for 1 h. The reaction mixture was filtered to remove tar material, and the filtrate was concentrated under reduced pressure to a volume of 100 mL and extracted with ethyl acetate (250 mL, twice). The extract was washed with saturated brine (100 mL), dried over anhydrous MgSO4, and concentrated to give crude 3a and c (16.3 g) as an amorphous substance. The amorphous substance was recrystallized from water, and the obtained crystals were dried under reduced pressure to give 3a and c (cis-isomers, 5.3 g, 22.7 mmol, 21.7%).
1H-NMR (DMSO-d6) δ: 1.80 (3H, s), 2.02 (3H, s), 3.49 (1H, dd, J=1.0, 12.0 Hz), 3.61 (1H, dd, J=6.3, 12.0 Hz), 5.22 (1H, dd, J=1.0, 6.2 Hz). 1H-NMR (D2O) δ: 1.80 (3H, s), 2.03 (3H, s), 3.41 (1H, d, J=12.4 Hz), 3.72 (1H, dd, J=6.4, 12.4 Hz), 5.05 (1H, d, J=6.4 Hz). 13C-NMR (D2O) δ: 22.8, 24.2, 34.1, 67.1, 73.7, 172.4, 174.7, 176.7. ESI-MS m/z: 234.0 (M+H)+, 232.0 (M−H)−. FAB-MS m/z: 234.0451 (M+H) (Calcd for C8H12NO5S: 234.0436).
N-Acetyl-2-methylthiazolidine-2,4-dicarboxylic Acid Anhydride (5a, c)Compounds 1a and b (2.0 g, 10.5 mmol) and acetic anhydride (4.0 mL, 42.0 mmol) were heated at 100°C for 2 h. The reaction mixture was then concentrated under reduced pressure. To the residue was added water (10 mL) and the residue was extracted with ethyl acetate (15 mL, twice). The extract was washed with saturated sodium chloride (10 mL, twice), dried over anhydrous MgSO4, and concentrated to give crude 5 (2.12 g) as an amorphous substance. The amorphous substance was purified by silica gel chromatography (eluted with n-hexane–ethyl acetate) to give 5a and c as a white solid (1.2 g, 5.6 mmol, 53.1%).
1H-NMR (DMSO-d6) δ: 1.78 (3H, s), 1.89 (3H, s), 3.63 (1H, dd, J=0.8, 12.6 Hz), 3.86 (1H, dd, J=5.8, 12.6 Hz), 4.91 (1H, dd, J=0.9, 5.7 Hz). ESI-MS m/z: 215.8 (M+H)+ (weak), 231.8 (M+H2O−H)−. Rf 0.80 (ethyl acetate–AcOEt (1 : 1), Kieselgel 60).
N-Acetyl-2-methylthiazolidine-2,4-dicarboxylic Acid 2-Ethyl Ester (trans-Isomers of AcCP2Et, 4d and b, Table 1, Entry 2)Compounds 2a and b (10.0 g, 45.6 mmol) was dissolved in ethyl acetate (100 mL), and the solution was kept at 0°C. To the solution was added Et3N (12.7 mL, 91.2 mmol), and acetyl chloride (4.86 mL, 68.4 mmol) was added dropwise over 10 min. The reaction temperature was allowed to rise gradually, and the mixture was stirred overnight at r.t. The reaction mixture was then concentrated under reduced pressure. To the residue was added ethyl acetate (200 mL), and the organic layer was washed with water (50 mL), aqueous citric acid (5%, 50 mL), aqueous sodium hydrogen carbonate (50 mL), and saturated sodium chloride (50 mL). The organic layer was dried over anhydrous MgSO4, and concentrated. The residue (trans–cis=95 : 5) was crystalized from ethyl acetate and n-heptane to give 4b and d (trans-isomer: 99%) as pale yellow crystals (7.0 g, 26.8 mmol, 58.8%).
1H-NMR (CDCl3) δ: 1.27 (3H, t, J=7.1 Hz), 1.94 (3H, s), 2.18 (3H, s), 3.40 (1H, d, J=11.6 Hz), 3.56 (1H, dd, J=5.5, 11.0 Hz), 4.20 (2H, qd, J=7.1, 7.2 Hz), 5.00 (1H, d, J=5.9 Hz), 9.10 (1H, br s). 1H-NMR (DMSO-d6) δ: 1.15 (3H, t, J=7.1 Hz), 1.77 (3H, s), 2.03 (3H, s), 3.36 (1H, dd, J=5.7, 11.6 Hz), 3.40 (1H, dd, J=10.6, 11.7 Hz), 4.04 (2H, qd, J=7.1, 9.0 Hz), 5.34 (1H, d, J=5.2 Hz). 13C-NMR (DMSO-d6, 100 MHz) δ: 14.3, 23.2, 23.9, 33.6, 61.4, 65.4, 70.7, 168.9, 171.4, 171.9. FAB-MS m/z: 262.0753 (M+H) (Calcd for C10H16NO5S: 262.0749). mp: 138–141°C.
N-Acetyl-2-methylthiazolidine-2,4-dicarboxylic Acid (trans-Isomers of AcCP, 3b and d, Table 1, Entry 2)Compounds 4b and d (20.0 g, 91.3 mmol) obtained from compounds 2a and b by an operation similar to that described above was dissolved in ethanol (40 mL), and sodium hydroxide solution (4 N, 57.0 mL) was added to the solution. The mixture was stirred with heating at 60°C under an argon atmosphere overnight. The mixture was allowed to cool to r.t., and the pH of the solution was adjusted to 1 with hydrochloric acid solution (6 N). The solution was kept at r.t. overnight, and the resulting white crystals were collected by filtration and washed with a small amount of cold water. The crystals were dried under reduced pressure to give 3b and d (trans-isomers, 14.2 g, 61.0 mmol, 66.8%).
1H-NMR (DMSO-d6) δ: 1.73 (3H, s), 2.01 (3H, s), 3.36 (2H, d, J=3.6 Hz), 5.28 (1H, t, J=3.6 Hz). 1H-NMR (D2O) δ: 1.77 (3H, s), 2.10 (3H, s), 3.43 (1H, d, J=12.0 Hz), 3.50 (1H, dd, J=6.0, 12.0 Hz), 5.24 (1H, d, J=5.6 Hz). 13C-NMR (D2O) δ: 22.1, 23.0, 32.9, 66.1, 70.8, 172.6, 173.6, 175.4. ESI-MS m/z: 234.0 (M+H)+, 232.0 (M−H)−. FAB-MS m/z: 234.0451 (M+H) (Calcd for C8H12NO5S: 234.0436).
N-Acetyl-2-methylthiazolidine-2,4-dicarboxylic Acid 2-Ethyl Ester (cis- and trans-Isomers of AcCP2Et, 4a and b, Table 1, Entry 4)Compounds 2a and b (20.0 g, 91.3 mmol) was dissolved in ethyl acetate (190 mL), and the solution was kept at 0°C. To the solution was added potassium carbonate (15.2 g, 110 mmol), and acetyl chloride (7.8 mL, 110 mmol) was added dropwise over 40 min. The reaction temperature was allowed to gradually rise, and the mixture was stirred overnight at r.t. To the solution was added hydrochloric acid aqueous solution (2N) until the pH of the aqueous layer was 2. The organic layer was separated and the water layer was extracted with ethyl acetate (200 mL, twice). The combined organic layers were washed with water (100 mL, twice) and saturated sodium chloride (100 mL, twice), dried over anhydrous MgSO4, and concentrated. The residue (cis–trans=50 : 50) was crystalized from ethyl acetate and n-hexane to give 4a and b (cis–trans=50 : 50) as colorless crystals (22.3 g, 85.5 mmol, 93.6%).
Pure trans-isomer 4b was obtained by recrystallization of the above mentioned diastereomeric mixture from ethyl acetate (150 mL) as colorless crystals (7.3 g). The cis-isomer was obtained by an operation similar to that outlined above, except that dichloromethane was used as the reaction and recrystallization solvent (36% yield, cis-isomer: 96%). The 1H-NMR and 13C-NMR spectra of trans-isomer 4b are virtually identical to those of the racemic mixture of 4b and d.
1H-NMR (CDCl3) δ: cis-isomer; 1.39 (3H, t, J=7.1 Hz), 1.99 (3H, s), 2.16 (3H, s), 3.47 (1H, dd, J=1.7, 12.2 Hz), 3.72 (1H, dd, J=6.8, 12.2 Hz), 4.33–4.48 (2H, m), 4.98 (1H, dd, J=1.7, 6.8 Hz). 13C-NMR (DMSO-d6, 100 MHz) δ: cis-isomer: 14.2, 24.1, 25.3, 33.0, 61.4, 65.1, 72.9, 168.8, 170.4, 170.8. FAB-MS m/z: 262.0753 (M+H) (Calcd for C10H16NO5S: 262.0749). mp: 101–105°C (cis-isomer).
N-Acetyl-2-methylthiazolidine-2,4-dicarboxylic Acid (cis-, trans-Isomers of AcCP, 3a, b, Table 1, Entry 4)Compounds 4a and b (5.0 g, 19.1 mmol) obtained from compounds 2a and b by an operation similar to that described above was dissolved in ethanol (10 mL), and sodium hydroxide solution (4 N, 12.0 mL) was added to the solution. The mixture was stirred with heating at 60°C under an argon atmosphere overnight. The mixture was allowed to cool to r.t., and the pH of the solution was adjusted to ca. 7 with an ion-exchange resin (Amberlite IR 120B H AG(H+)), filtered and concentrated under reduced pressure to give 3a, b and small amount of 3d as a colorless amorphous (4.4 g, 18.9 mmol, 99.5%).
N-Acetyl-2-methylthiazolidine-2,4-dicarboxylic Acid 2,4-Diethyl Ester (10a, Chart 3)Compounds 4a and b (cis–trans=48 : 52, 1.0 g, 3.84 mmol) was dissolved in dichloromethane (15 mL), and the reaction mixture was kept at 0°C. To the solution were added ethanol (0.27 mL, 4.60 mmol), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDCI·HCl, 0.89 g, 4.64 mmol), and 4-dimethylaminopyridine (DMAP; 95 mg, 0.77 mmol). The reaction temperature was allowed to gradually rise to r.t., and the mixture was stirred overnight. The reaction mixture was concentrated under reduced pressure, and water (10 mL) was added. The mixture was then extracted with ethyl acetate (10 mL, three times). The combined organic layer was washed with aqueous citric acid (5%, 10 mL, twice), aqueous sodium hydrogen carbonate (5%, 10 mL, twice) and saturated sodium chloride (10 mL), dried over anhydrous MgSO4, and concentrated to give 10a as a colorless oil (cis–trans=45 : 55, 1.0 g, 3.48 mmol, 91.0%).
1H-NMR (CDCl3) δ: cis-isomer; 1.28 (3H, t, J=7.1 Hz), 1.34 (3H, t, J=7.1 Hz), 1.96 (3H, s), 2.17 (3H, s), 3.50 (1H, dd, J=6.1, 11.8 Hz), 3.61 (1H, dd, J=1.8, 11.9 Hz), 4.26–4.21 (2H, m), 4.34–4.27 (2H, m), 4.85 (1H, dd, J=1.8, 6.0 Hz). trans-Isomer; 1.27 (3H, t, J=7.1 Hz), 1.33 (3H, t, J=7.2 Hz), 1.93 (3H, s), 2.12 (3H, s), 3.36 (1H, d, J=11.7 Hz), 3.54 (1H, dd, J=6.2, 11.6 Hz), 4.12–4.25 (2H, m), 4.25–4.33 (2H, m), 4.95 (1H, d, J=5.84 Hz). FAB-MS m/z: 290.1063 (M+H) (Calcd for C12H20NO5S: 290.1062).
N-Acetyl-2-methylthiazolidine-2,4-dicarboxylic Acid 2-Ethyl 4-Isopropyl Diester (10b, Chart 3)An operation similar to the one described for 10a was performed using isopropanol, and 10b was obtained in an overall yield of 86% as a colorless oil (cis–trans=27 : 73).
1H-NMR (CDCl3) δ: cis-isomer; 1.28 (3H, t, J=7.1 Hz), 1.33 (6H, d, J=6.2 Hz), 1.96 (3H, s), 2.16 (3H, s), 3.49 (1H, dd, J=6.1, 11.9 Hz), 3.59 (1H, dd, J=2.0, 1.9 Hz), 4.81 (1H, dd, J=2.0, 6.1 Hz), 5.19–5.12 (1H, m). trans-Isomer; 1.28 (3H, t, J=7.1 Hz), 1.31 (6H, d, J=6.2 Hz), 1.93 (3H, s), 2.12 (3H, s), 3.36 (1H, dd, J=0.7, 11.6 Hz), 3.55 (1H, dd, J=6.2, 11.6 Hz), 4.17–4.28 (2H, m), 4.91 (1H, d, J=5.6 Hz), 5.12–5.19 (1H, m). FAB-MS m/z: 304.1208 (M+H) (Calcd for C13H22NO5S: 304.1219).
N-Acetyl-2-methylthiazolidine-2,4-dicarboxylic Acid 2-Ethyl Ester 4-Gly Methyl Ester Amide (13a, Chart 3)Compounds 4a and b (cis–trans=48 : 52, 2.0 g, 7.68 mmol) was dissolved in dichloromethane (20 mL), and the reaction mixture was kept at 0°C. To the solution were added Gly methyl ester hydrochloride (0.963 g, 7.67 mmol), EDCI·HCl (1.62 g, 8.47 mmol) and 1-hydroxy-1H-benzotriazole hydrate (HOBt·H2O, 1.14 g, 8.45 mmol). The reaction temperature was allowed to gradually rise to r.t., and the mixture was stirred overnight. The reaction mixture was concentrated under reduced pressure, and water (20 mL) was added. The mixture was then extracted with ethyl acetate (80 mL, three times). The combined organic layer was washed with aqueous citric acid (5%, 20 mL, twice), aqueous sodium hydrogen carbonate (5%, 20 mL, twice) and saturated sodium chloride (20 mL), dried over anhydrous MgSO4, and concentrated to give 13a as a pale-yellow oil (cis–trans=48 : 52, 2.5 g, 7.20 mmol, 93.8%).
1H-NMR (CDCl3) δ: cis-isomer; 1.30 (3H, t, J=7.1 Hz), 2.07 (3H, s), 2.21 (3H, s), 3.36 (1H, d, J=11.9 Hz), 3.62 (1H, dd, J=7.0, 11.9 Hz), 4.35–4.14 (4H, m), 3.81(3H, s), 4.90 (1H, d, J=6.8 Hz), 6.89 (1H, br s). trans-Isomer; 1.36 (3H, t, J=7.1 Hz), 1.99 (3H, s), 2.23 (3H, s), 3.44 (1H, dd, J=1.1, 12.3 Hz), 3.70 (1H, dd, J=7.0, 12.3 Hz), 3.76 (3H, s), 4.35–4.14 (4H, m), 4.84 (1H, dd, J=1.0, 7.0 Hz), 9.09 (1H, br s). FAB-MS m/z: 333.1118 (M+H) (Calcd for C13H21N2O6S: 333.1120).
N-Acetyl-2-methylthiazolidine-2,4-dicarboxylic Acid 2-Ethyl Ester 4-L-Ala Methyl Ester Amide (13b, Chart 3)An operation similar to the one described for 13a was performed using L-Ala methyl ester hydrochloride, and 13b was obtained in quantitative yield of as a pale-yellow oil (cis–trans=48 : 52).
1H-NMR (CDCl3) δ: cis-isomer; 1.29 (3H, t, J=7.1 Hz), 1.48 (3H, d, J=7.4), 2.02 (3H, s), 2.21 (3H, s), 3.35 (1H, d, J=11.9 Hz), 3.60 (1H, dd, J=6.9, 11.9 Hz), 3.78 (3H, s), 4.30–4.36 (2H, m), 4.85 (1H, d, J=6.6 Hz), 6.83 (1H, br s), 9.00 (1H, br s). trans-Isomer; 1.35 (3H, t, J=7.1 Hz), 1.48 (3H, d, J=7.2 Hz), 1.99 (3H, s), 2.24 (3H, s), 3.41 (1H, dd, J=0.9, 12.2 Hz), 3.67 (1H, dd, J=7.0, 12.2 Hz), 3.74 (3H, s), 4.19–4.24 (2H, m), 4.62–4.68 (1H, m), 4.52–4.60 (1H, m), 4.80 (1H, dd, J=0.8, 6.9 Hz), 9.00 (1H, br s). FAB-MS m/z: 347.1285 (M+H) (Calcd for C14H23N2O6S: 347.1277).
N-Hexadecanoyl-2-methylthiazolidine-2,4-dicarboxylic Acid 2-Ethyl Ester (trans-Isomer, 11, Chart 3)Compounds 2a and b (trans–cis=50 : 50, 10.0 g, 45.6 mmol) was dissolved in ethyl acetate (90 mL), and the solution was kept at 0°C. To the solution was added Et3N (12.7 mL, 91.2 mmol), and n-hexadecanoyl chloride (20.7 mL, 68.4 mmol) was added dropwise over 30 min. The reaction temperature was allowed to gradually rise to r.t., and the mixture was stirred overnight. Citric acid solution (5%, 30 mL) was then added to the reaction mixture, and the organic layer was separated. The aqueous layer was extracted with ethyl acetate (60 mL, twice). The combined organic layer was washed with water (50 mL, twice) and saturated sodium chloride (50 mL, twice), dried over anhydrous MgSO4, and concentrated. The residue was purified by silica gel chromatography (eluted with n-hexane–ethyl acetate=9 : 1 to 1 : 1, 1% AcOH) to give a pale yellow solid. The obtained solid was washed with n-hexane to give 11 as a pale yellow wax (trans-isomer, 13.2 g, 28.8 mmol, 63.2%).
1H-NMR (CDCl3) δ: 0.90 (3H, t, J=6.9 Hz), 1.28 (3H, t, J=7.1 Hz), 1.27–1.36 (24H, m), 1.62–1.67 (2H, m), 1.96 (3H, s), 2.27–2.33 (2H, m), 3.40 (1H, d, J=11.6 Hz), 3.59 (1H, dd, J=6.27, 11.8 Hz), 4.18–4.28 (2H, m), 5.06 (1H, d, J=6.0 Hz). ESI-MS m/z: 458.4 (M+H)+, 456.3 (M−H)−. FAB-MS m/z: 458.2950 (M+H) (Calcd for C24H44NO5S: 458.2940).
N-Hexadecanoyl-2-methylthiazolidine-2,4-dicarboxylic Acid (trans-Isomer, 12, Chart 3)To the solution of compound 11 (trans-isomer, 5.12 g, 11.2 mmol) in EtOH (10 mL) was added aqueous NaOH solution (4 N, 6.97 mL, 27.88 mmol), and the solution was stirred overnight at 60°C. The pH of the solution was adjusted to 1 by addition of aqueous HCl (6N). The solution was then extracted with ethyl acetate (30 mL, twice) and the combined organic layer was washed with water (30 mL, twice) and saturated sodium chloride (30 mL, twice), dried over anhydrous MgSO4, and concentrated. The residue was recrystallized from mixed solvent of ethyl acetate and n-hexane to give 12 as a white solid (trans-isomer, 4.16 g, 9.68 mmol, 86.4%).
1H-NMR(400 MHz, CDCl3) δ: 0.88 (3H, t, J=6.9 Hz), 1.23–1.29 (24H, m), 1.62–1.67 (2H, m), 1.94 (3H, s), 2.25–2.36 (2H, m), 3.42 (1H, d, J=11.2 Hz), 3.59 (1H, dd, J=6.1, 11.9 Hz), 5.04 (1H, d, J=6.7 Hz). ESI-MS m/z: 430.3 (M+H)+, 428.3 (M−H)−. FAB-MS m/z: 430.2620 (M+H) (Calcd for C22H40NO5S: 430.2627).
2-Methylthiazolidine-2,4-dicarboxylic Acid 2-Methyl Ester (CP2Mt, 8, Chart 3)An operation similar to the one described for compounds 2a and b was performed using methyl pyruvate, and 8 was obtained in an overall yield of 71.5% as a white solid (ca. 50 : 50 diastereomer mixture).
1H-NMR(DMSO-d6) δ: diastereomer 1; 1.74 (3H, s), 2.96 (1H, dd, J=8.7, 10.0 Hz), 3.27 (1H, dd, J=6.7, 10.0 Hz), 3.62 (3H, s), 4.13 (1H, d, J=6.8, 8.5 Hz). Diastereomer 2; 1.60 (3H, s), 1.60 (3H, s), 3.41 (1H, dd, J=6.1, 10.3 Hz), 3.72 (3H, s), 4.01 (1H, dd, J=6.1, 9.9 Hz).
N-Acetyl-2-methylthiazolidine-2,4-dicarboxylic Acid 2-Methyl Ester (AcCP2Me, 9, Chart 3)An operation similar to the one described for compounds 4a and b was performed using compounds 8 and 9 was obtained in an overall yield of 85.2% (cis–trans=50 : 50) as colorless crystals (10.3 g, 41.5 mmol). The pure cis-isomer and trans-isomer were obtained by repeated recrystallization of the diastereomeric mixture from ethyl acetate.
1H-NMR (CDCl3) δ: cis-isomer; 2.01 (3H, s), 2.17 (3H, s), 3.49 (1H, dd, J=1.7, 12.3 Hz), 3.72 (1H, dd, J=6.7, 12.2 Hz), 3.95 (3H, s), 5.00 (1H, dd, J=1.7, 6.6 Hz). trans-Isomer; 1.95 (3H, s), 2.19 (3H, s), 3.49 (1H, d, J=11.6 Hz), 3.58 (1H, dd, J=6.7, 12.2 Hz), 3.76 (3H, s), 5.01 (1H, d, J=5.8 Hz). FAB-MS m/z: 248.0607 (M+H) (Calcd for C9H14NO5S: 248.0593).
Single Crystal X-Ray StudySingle crystals of the mixture of cis-(2R,4R)- and cis-(2S,4S)-isomers of AcCP (3a, c) (Table 1, entry 1) and the mixture of trans-(2S,4R)- and trans-(2R,4S)-isomers of AcCP (3b, d) (Table 1, entry 2) suitable for X-ray analysis were obtained from MeOH via slow evaporation.
X-Ray diffraction (XRD) data were collected using a Rigaku RAXIS RAPID imaging plate area detector with filtered CuKα radiation (λ=1.54187 Å) at a temperature of 20±1°C. The crystal-to-detector distance was 127.40 mm. Readout was performed in the 0.100 mm pixel mode. The exposure rate was set to 2.0 s/°. The XRD data of each crystal were recorded to a total of 45 oscillation images. A first sweep of data was performed using ω scans from 80.0 to 260.0° in 20.0° steps at χ=0.0° and ϕ=0.0°. A second sweep was performed using ω scans from 80.0 to 260.0° in 20.0° steps at χ=54.0° and ϕ=0.0°. A third sweep was performed using ω scans from 80.0 to 260.0° in 20.0° steps at χ=54.0° and ϕ=90.0°. A fourth sweep was performed using ω scans from 80.0 to 260.0° in 20.0° steps at χ=54.0° and ϕ=180.0°. A fifth sweep was performed using ω scans from 80.0 to 260.0° in 20.0° steps at χ=54.0° and ϕ=270.0°. An empirical absorption correction was applied to the data. The data were also corrected for Lorentz and polarization effects and the secondary extinction effect.
Crystal structures were solved using direct methods26) and expanded using Fourier techniques.27) Furthermore, crystallographic refinement of full-matrix least-squares was performed. The non-hydrogen atoms were refined anisotropically. Some hydrogen atoms were refined isotropically, some were refined using the riding model, and the rest were included in fixed positions. In the final cycles of the refinement, a least-squares function of ∑ w(Fo2−Fc2)2 was adopted, where w is the least-squares weights based on the Sheldrick weighting scheme. All calculations were performed using the CrystalStructure crystallographic software package.28,29)
Suppression of Eumelanin Production TestingB16 melanoma cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM; high glucose) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin (10000 U/mL)–streptomycin (10 mg/mL) solution. Cells were incubated in a humidified 5% CO2 atmosphere at 37°C.
B16 melanoma cells were plated in 96-well microplates at 8.0×103 cells per well in 200 µL of DMEM containing 10% FBS without phenol red. After overnight incubation, the medium was replaced with 200 µL of DMEM containing each sample at a predetermined concentration. Cells were treated with a predetermined concentration of each sample for 3 d. The absorbance of the medium at 450 nm after 3 d was measured by a microplate reader.
The concentration necessary for suppressing eumelanin production in each sample by 50% was calculated relative to the amount of eumelanin in the control as 100%. Cytotoxicity was determined by a neutral red assay as follows: After incubation for 3 d following the addition of each sample, the cells were incubated with neutral red dye (50 µg/mL) for 2 h and subsequently fixed with 1% formaldehyde and 1% CaCl2 for 1 min. The dye was extracted with the solvent solution (1% acetic acid in 50% ethyl alcohol) and was quantitated by measuring the absorbance at 540 nm in a microplate reader. The absorbance is given as a relative percentage of the absorbance of the control as 100%.
The absorbance after 3 d following the addition of a predetermined concentration of each sample is shown as a relative percentage of the absorbance of the control as 100%.
Time–Course Stability Test of AcCP2Et (4a, b) at 70°CA time–course stability test was performed using a ca. 1 : 1 mixture (20 mg/20 mL) of cis- and trans-isomers of AcCP2Et (4a, b) (Table 1, entry 4) in a 25 mM phosphate buffer at pH 4, 5, 6, and 7 at 70°C. The test was performed over 5 d. As an index, the residual ratio was calculated from the RP-HPLC area value of a target substance after 5 d divided by that on day 0. The results are shown in Table 2.
Odor TestAqueous solutions (1%) at pH 4 and 7 of trans-enantiomers of AcCP2Et (4b, d) (Table 1, entry 2) and CP2Et (2a, b) were prepared, placed in tightly sealed vials, and preserved in a thermostatic bath at 70°C for 6 d. Six panelists then evaluated the odor of the samples according to the following criteria; 3 points: no sulfur odor was detected at all, 2 points: a slight sulfur odor was detected, 1 point: sulfur odor was detected, and 0 point: strong sulfur odor was detected. The results of the 6 panelists were totaled as follows; AcCP2Et (4b, d) at pH 4: 8 points, AcCP2Et (4b, d) at pH 7: 17 points, CP2Et (2a, b) at pH 4: 1 point, CP2Et (2a, b) at pH 7: 1 point.