Experimental
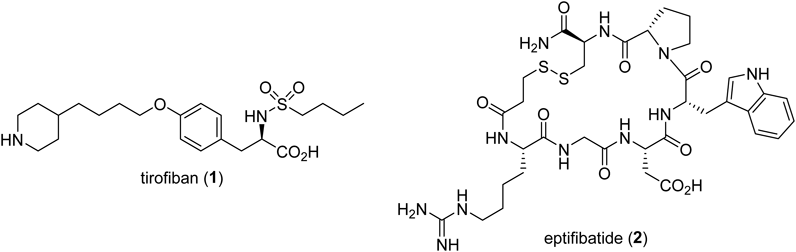
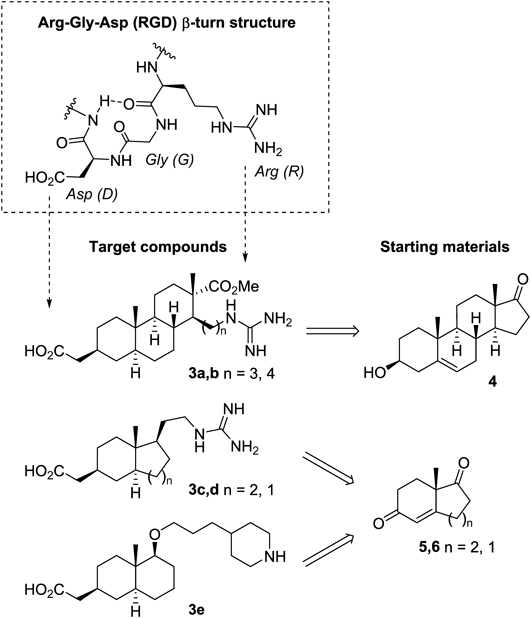
General MethodsAll melting points were measured on a Yanagimoto micro melting point apparatus and are uncorrected. Optical rotations were obtained using a JASCO P-2200 polarimeter. Optical purities were determined on a JASCO HPLC (PU-2089 plus, MD-2015 plus) instrument equipped with Daicel Chemical chiral columns. IR spectra were recorded on a Shimadzu IRPrestige-21 spectrophotometer. 1H- and 13C-NMR spectra were measured on a JEOL JNM-AL300 (300 MHz), JEOL JMN-AL400 (400 MHz), JEOL JNM-LA500 (500 MHz) or JNM-ECA500 (500 MHz) spectrometers with tetramethylsilane as an internal standard. J-Values are given in Hertz. Mass spectra were recorded on a JEOL JMS-DX302 or JEOL JMS 700 instruments with a direct inlet system. Elemental analysis was performed using a Yanaco MT-6 elemental analyzer. Column chromatography was carried out on silica gel [Kanto Chemical Co., Inc., Japan (Silica Gel 60N, Spherical, neutral 40–50 µm) and Merck Ltd. (Silica Gel 60F254, 230–400 mesh)]. TLC was carried out on silica gel plates [Merck Ltd., Germany (TLC Silica gel 60F254)] and visualized with UV254 light and/or staining with phosphomolybdic acid in EtOH. The purity of compounds 3a–e were measured by the internal standard method using 1,4-bis(trimethylsilyl)benzene.38)
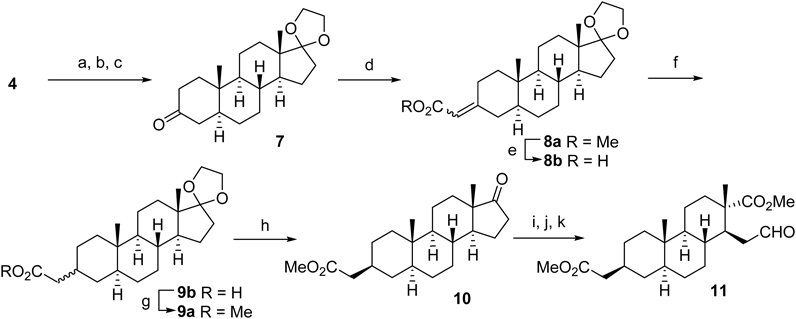
(3S,8R,9S,10R,13S,14S)-10,13-Dimethyl-1,2,3,4,7,8,9,10,11,12,13,14,15,16-tetradecahydrospiro[cyclopenta[a]phenanthrene-17,2′-[1,3]dioxolan]-3-olTo a solution of 4 (25.0 g, 86.7 mmol) in benzene (420 mL) were added ethylene glycol (14.5 mL, d 1.11, 259 mmol) and camphorsulfonic acid (4.0 g, 17.2 mmol). The reaction mixture was heated under reflux using Dean-Stark apparatus for 3 h. The mixture was cooled to room temperature (r.t.) and neutralized using satd aqueous NaHCO3 on an ice cooling bath. The mixture was extracted with EtOAc and the combined organic layer was washed with brine. The extract was dried over MgSO4 and concentrated under reduced pressure. The residue was purified using column chromatography (AcOEt–n-hexane=1 : 8) and acetal (23 g, 80%) was obtained as a white powder.
mp 166–168°C (AcOEt–n-hexane); [α]D27 −92.3 (c=0.48, CHCl3); Anal. Calcd for C21H32O3: C, 75.86; H, 9.70. Found: C, 75.82; H, 9.60.
(8R,9S,10R,13S,14S)-10,13-Dimethyl-1,6,7,8,9,10,11,12,13,14,15,16-dodecahydrospiro[cyclopenta[a]phenanthrene-17,2′-[1,3]dioxolan]-3(2H)-oneUnder nitrogen atmosphere, Al(Oi-Pr)3 (2.6 g, 12.7 mmol) and cyclohexanone (39.7 mL, d 0.95, 383 mmol) were added to a toluene (255 mL) solution of acetal (8.5 g, 25.5 mmol) at r.t. After the 2 h reflux, the reaction mixture was cooled to r.t. and added satd aqueous potassium sodium tartrate. The aqueous layer was extracted with AcOEt and the combined extract was dried over MgSO4. The concentrated residue was purified by column chromatography (AcOEt–n-hexane=1 : 3) to give enone (8.2 g, 97%) as colorless crystals.
mp 148–149°C (AcOEt–n-hexane); [α]D27 +62.2 (c=0.49, CHCl3); Anal. Calcd for C21H30O3: C, 76.33; H, 9.15. Found: C, 76.17; H, 8.98.
(5S,8R,9S,10S,13S,14S)-10,13-Dimethyltetradecahydrospiro[cyclopenta[a]phenanthrene-17,2′-[1,3]dioxolan]-3(2H)-one (7)Under a nitrogen atmosphere, a lithium wire (1.0 g, 144 mmol) was added to liquid ammonia (300 mL) at −78°C. To the reaction mixture was added the solution of enone (4.0 g, 12.1 mmol) in tetrahydrofuran (THF) (150 mL) and stirred for 30 min. After the substrate disappearance was monitored by TLC, solid NH4Cl was added until the mixture turned white suspension. The mixture was allowed to r.t. and the liquid ammonia was evaporated through the night. The residue was suspended with CH2Cl2 and filtrated. The concentrated residue was purified by column chromatography (AcOEt–n-hexane=1 : 3) to give compound 7 (3.5 g, 88%) as white crystals.
mp 203–204°C (MeOH); [α]D28 +2.1 (c=0.47, CHCl3); Anal. Calcd for C21H32O3: C, 75.86; H, 9.70. Found: C, 75.96; H, 9.91.
Methyl 2-((5S,8R,9S,10S,13S,14S)-10,13-Dimethyltetradecahydrospiro[cyclopenta[a]phenanthrene-17,2′-[1,3]dioxolan]-3(2H)-ylidene)acetate (8a)To a suspension of t-BuOK (5.3 g, 47.5 mmol) in anhydrous DMF (95 mL) was added methyl diethylphosphonoacetate (10.5 mL, d 1.15, 57.0 mmol) at r.t. and stirred for 1 h under a nitrogen atmosphere. The mixture was cooled to 0°C and added the solution of 7 (6.3 g, 19.0 mmol) in anhydrous DMF (95 mL) and stirred for 12.5 h. The reaction mixture was poured into water and extracted with Et2O. The combined extracts were washed with brine, dried over MgSO4 and concentrated under the reduced pressure. The residue was purified by column chromatography (AcOEt–n-hexane=1 : 6) to give compound 8a (7.1 g, 97%) as a white powder.
mp 199–201°C (MeOH); [α]D28 +68.8 (c=0.16, CHCl3); IR (CHCl3) cm−1: 2945, 1705, 1643, 1204, 1165; 1H-NMR (CDCl3, 400 MHz) δ: 5.59 (t, 1H, J=1.6 Hz), 4.04–3.80 (m, 4H), 3.67 (s, 3H), 2.44–2.26 (m, 1H), 2.22–2.08 (m, 1H), 2.06–0.45 (m, 20H), 0.94 (s, 3H), 0.85 (s, 3H); 13C-NMR (CDCl3, 100 MHz) δ: 167.0, 163.5, 119.3, 112.1, 65.2, 64.6, 54.1, 50.8, 50.4, 47.6, 46.0, 40.2, 36.2, 35.8, 34.3, 33.7, 32.3, 31.4, 30.8, 29.0, 22.8, 20.7, 14.6, 12.1; HR-MS (EI) Calcd for C24H36O4 m/z [M+]: 388.2614. Found 388.2612; Anal. Calcd for C24H36O4: C, 74.19; H, 9.34. Found: C, 74.04; H, 9.59.
Methyl 2-((3S,5S,8R,9S,10S,13S,14S)-10,13-Dimethyl-17-oxohexadecahydro-1H-cyclopenta[a]phenanthren-3-yl)acetate (10)To a suspension of 8a (7.1 g, 18.3 mmol) in 1 : 1 EtOH/H2O (184 mL) was added KOH (10.3 g, 183 mmol) and the mixture was heated at 90°C for 1.5 h. The reaction mixture was concentrated and neutralized with dilute HCl on the ice bath. The mixture was extracted with CH2Cl2, and the combined organic layer was dried over MgSO4. The filtrate was concentrated to obtain the crude α,β-unsaturated carboxylic acid 8b.
Under the nitrogen atmosphere, a lithium wire (1.3 g, 187 mmol) was added to liquid ammonia (30 mL) at −78°C. To the mixture was added the solution of the above crude α,β-unsaturated carboxylic acid 8b in 1,4-dioxane (95 mL), THF (95 mL) and t-BuOH (7.1 mL, d 0.78, 74.2 mmol). After 3 h stirring, MeOH was added until the mixture turned white suspension. The mixture was allowed to r.t. and the liquid ammonia was evaporated through the night. The residue was suspended with CH2Cl2 and filtrated. The solution was acidified using dilute HCl and the water layer was extracted by CH2Cl2. The combined organic layer was dried over MgSO4 and the concentrated residue was evaporated to give the crude carboxylic acid 9b. The ratio of isomers was determined the benzyl esters of 9b by 1H-NMR.
Under the nitrogen atmosphere, sodium hydride (NaH) (2.2 g, 60% oil suspension, 54.9 mmol) was added to the above crude carboxylic acid 9b in anhydrous DMF (183 mL) at r.t. After the reaction mixture was stirred for 1 h, methyl iodide (MeI) (8.0 mL, 54.9 mmol) was added and the reaction mixture was stirred overnight. The reaction was concentrated, diluted by Et2O and washed with H2O. The organic layer was dried over MgSO4 and concentrated to give crude methyl ester.
To the above crude methyl ester in THF (183 mL) was added 1.5 N HCl (245 mL) using the dropping funnel at 0°C and the mixture was stirred for 4 h at r.t. The reaction mixture was neutralized with 10% NaOH and concentrated under reduced pressure. The residue was extracted with AcOEt and the organic layer was dried over MgSO4. The concentrated residue was purified by column chromatography (AcOEt–n-hexane=1 : 7) to give compound 10 (5.8 g, 91%, 4 steps) as a white powder.
mp 110–115°C (AcOEt–n-hexane); [α]D28 +74.3 (c=0.50, CHCl3); IR (CHCl3) cm−1: 3012, 2920, 2855, 1728, 1439; 1H-NMR (CDCl3, 400 MHz) δ: 3.64 (s, 3H), 2.41 (dd, 1H, J=19.0, 8.7 Hz), 2.18 (d, 2H, J=7.1 Hz), 1.91 (dt, 1H, J=19.0, 9.0 Hz), 1.97–1.86 (m, 1H), 1.86–0.60 (m, 20H), 0.84 (s, 3H), 0.77 (s, 3H); 13C-NMR (CDCl3, 100 MHz) δ: 220.9, 173.2, 54.6, 51.5, 51.4, 47.8, 46.4, 41.8, 38.3, 36.0, 35.9, 35.24, 35.21, 35.1, 31.7, 31.0, 28.59, 28.57, 21.9, 20.4, 13.9, 12.4; HR-MS (EI) Calcd for C22H34O3 m/z [M+]: 346.2508. Found 346.2511.
Methyl 2-((3S,5S,8R,9S,10S,13S,14S)-17-Acetoxy-10,13-dimethyl-2,3,4,5,6,7,8,9,10,11,12,13,14,15-tetradecahydro-1H-cyclopenta[a]phenanthren-3-yl)acetateTo a solution of 10 (6.1 g, 17.6 mmol) in isopropenyl acetate (38.4 mL) was added the solution of conc. H2SO4 (0.1 mL) in isopropenyl acetate (5.0 mL) under a nitrogen atmosphere. After stirring for 5.5 h at 90°C, above H2SO4 in isopropenyl acetate (1.9 mL) was added and then the reaction mixture was stirred for 4.5 h at same temperature. The reaction mixture was diluted with Et2O and neutralized with satd aqueous NaHCO3 at 0°C. The mixture was extracted with Et2O and the combined organic layer was dried over MgSO4. The concentrated residue was purified by column chromatography (AcOEt–n-hexane=1 : 15) to give acetate (5.2 g, 75%) as a white solid.
IR (CHCl3) cm−1: 2920, 2855, 1732, 1229, 1202; 1H-NMR (CDCl3, 400 MHz) δ: 5.44 (dd, 1H, J=2.9, 1.5 Hz), 3.65 (s, 3H), 2.29–2.07 (m, 1H), 2.19 (d, 2H, J=7.1 Hz), 2.13 (s, 3H), 1.98–0.67 (m, 20H), 0.87 (s, 3H), 0.77 (s, 3H); 13C-NMR (CDCl3, 100 MHz) δ: 173.2, 168.6, 159.5, 111.1, 55.1, 54.2, 51.4, 46.7, 44.9, 41.9, 38.1, 36.1, 35.3 (CH), 35.3 (CH2), 33.7, 33.5, 31.2, 29.0, 28.7, 28.6, 21.3, 20.6, 15.7, 12.4; HR-MS (EI) Calcd for C24H36O4 m/z [M+]: 388.2614. Found 388.2617.
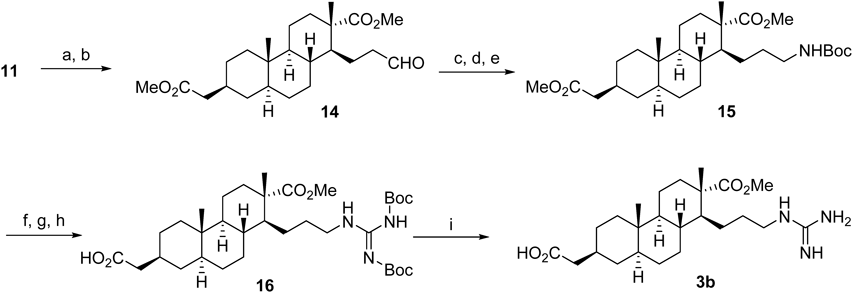
Methyl (1S,2S,4aS,4bS,7S,8aS,10aR)-7-(2-Methoxy-2-oxoethyl)-2,4b-dimethyl-1-(2-oxoethyl)tetradecahydrophenanthrene-2-carboxylate (11)To a solution of acetate (1.0 g, 2.6 mmol) and AcOH (2.1 mL) in CH2Cl2 (26 mL) was bubbled with ozone at −78°C for 3 h. The dissolved ozone gas was removed by Ar bubbling through the solution. Then Me2S (3.0 mL, d 0.85, 41.0 mmol) was added to the mixture, and the resulting mixture was stirred at 0°C for 6.5 h. After addition of AcOH (24 mL) and H2O (8 mL), the mixture was stirred at r.t. for 1 d. The reaction mixture was added H2O and extracted with CH2Cl2. The organic layer was dried over MgSO4 and concentrated in vacuo. The residue was diluted with Et2O, and a solution of CH2N2 in Et2O was added until the resulting solution turned to yellow. The mixture was warmed to r.t. and stirred overnight. The concentrated residue was purified by column chromatography (Et2O–n-hexane=1 : 5) to give compound 11 (845 mg, 83% in 2 steps) as a white solid.
mp 68–70°C (AcOEt–n-hexane); IR (CHCl3) cm−1: 3026, 2949, 2855, 1721, 1435; [α]D27 −26.6 (c=0.36, CHCl3); 1H-NMR (CDCl3, 400 MHz) δ: 9.68 (d, 1H, J=3.2 Hz), 3.654 (s, 3H), 3.646 (s, 3H), 2.42–2.31 (m, 1H), 2.27 (ddd, 1H, J=17.2, 7.2, 3.2 Hz), 2.19 (d, 2H, J=6.8 Hz), 2.10 (dd, 1H, J=17.2, 2.2 Hz), 1.88–0.67 (m, 18H), 1.11 (s, 3H), 0.72 (s, 3H). 13C-NMR (CDCl3, 100 MHz) δ: 201.9, 178.1, 173.2, 53.3, 52.0, 51.4, 47.4, 46.7, 45.8, 41.8, 41.7, 38.1, 37.4, 36.7, 35.9, 35.2, 35.0, 31.9, 28.7, 28.5, 19.8, 15.6, 12.3; HR-MS (FAB) Calcd for C23H37O5 m/z [MH+]: 393.2641. Found 393.2641.
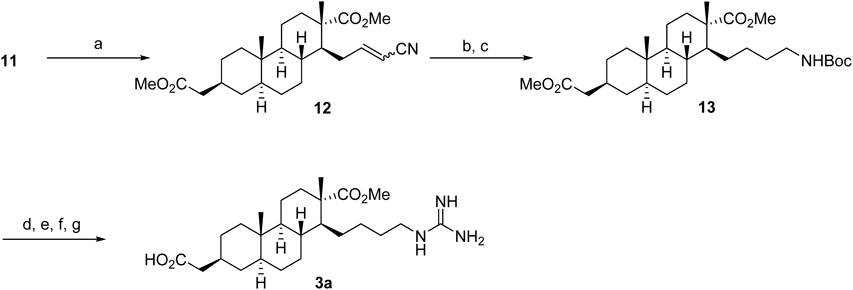
Methyl (1S,2S,4aS,4bS,7S,8aS,10aR)-1-(3-Cyanoallyl)-7-(2-methoxy-2-oxoethyl)-2,4b-dimethyltetradecahydrophenanthrene-2-carboxylate (12)To a suspension of KOtBu (44.0 mg, 0.38 mmol) in DMF (640 µL) was added diethyl cyanomethylphosphonate (59.6 µL, d 1.13, 0.38 mmol) and the mixture was stirred at 0°C for 1 h under a nitrogen atmosphere. To the solution was added compound 11 (50.0 mg, 0.13 mmol) in anhydrous DMF (640 µL) and stirred at r.t. for 10 h. The reaction mixture was poured into H2O and extracted with Et2O. The combined extract was dried over MgSO4 and concentrated. The residue was purified by column chromatography (Et2O–n-hexane=1 : 3) to give compound 12 (50.5 mg, 96%) as white amorphous.
IR (CHCl3) cm−1: 2949, 2922, 2224, 1721, 1630, 1435, 1236; [α]D27 −28.2 (c=0.24, CHCl3); 1H-NMR (CDCl3, 300 MHz, E : Z=1 : 1.8) δ: 6.70 (dt, 0.36H, J=16.5, 7.2 Hz), 6.49 (ddd, 0.64H, J=10.9, 6.8, 6.6 Hz), 5.28 (dt, 0.36H, J=16.5, 1.7 Hz), 5.21 (dt, 0.64H, J=10.9, 1.7 Hz), 3.670 (s, 1.1H), 3.665 (s, 1.9H), 3.663 (s, 1.9H), 3.661 (s, 1.1H), 2.51–2.30 (m, 0.64H), 2.26–2.09 (m, 1H), 2.20 (d, 2H, J=7.0 Hz), 2.08–1.95 (m, 0.36H), 1.91–0.73 (m, 19H), 1.15 (s, 1.9H), 1.10 (s, 1.1H), 0.74 (s, 1.9H), 0.73 (s, 1.1H); 13C-NMR (CDCl3, 100 MHz, E : Z=1 : 5.5) δ: 178.2, 172.9, 155.7, 115.7, 98.2, 53.3, 51.7, 51.2, 47.4, 46.9, 45.6, 41.6, 38.0, 37.3, 37.2, 35.8, 35.0, 34.9, 34.3, 31.5, 28.6, 28.3, 19.6, 15.2, 12.2; HR-MS (FAB) Calcd for C25H38NO4 m/z [MH+]: 416.2801. Found 416.2806.
Methyl (1S,2S,4aS,4bS,7S,8aS,10aR)-1-(4-((tert-Butoxycarbonyl)amino)butyl)-7-(2-methoxy-2-oxoethyl)-2,4b-dimethyltetradecahydrophenanthrene-2-carboxylate (13)Under a hydrogen atmosphere (10 atm), the suspension of compound 12 (144 mg, 0.35 mmol), Pt2O (27 mg) and 10% HCl–MeOH (9.0 mL) in CHCl3 (6.0 mL) was stirred at for 11.5 h in an autoclave at r.t. The reaction mixture was filtered through a Celite pad and the filtrate was concentrated. The residue was dissolved in CH2Cl2 (4.0 mL) and added Et3N (146 µL, d 0.73, 1.05 mmol) and Boc2O (161 µL, d 0.95, 0.70 mmol). After 7 h stirring, the reaction mixture was washed with H2O and dried over MgSO4. The concentrated residue was purified by column chromatography (AcOEt–n-hexane=1 : 5) to give compound 13 (174 mg, 95%, 2 steps) as a pale yellow oil.
[α]D28 −16.7 (c=0.46, CHCl3); IR (CHCl3) cm−1: 3453, 3024, 2928, 2857, 2359, 1715, 1506; 1H-NMR (CDCl3, 300 MHz) δ: 4.49 (br, 1H), 3.64 (s, 3H), 3.63 (s, 3H), 3.03 (dd, 2H, J=11.8, 6.3 Hz), 2.18 (d, 2H, J=6.9 Hz), 2.00–0.50 (m, 25H), 1.42 (s, 9H), 1.05 (s, 3H), 0.70 (s, 3H); 13C-NMR (CDCl3, 100 MHz) δ: 179.2, 173.5, 155.9, 78.9, 53.3, 51.5, 51.3, 47.7, 47.5, 45.8, 41.7, 40.3, 38.0, 37.7, 36.9, 35.8, 35.1, 35.0, 31.8, 31.6, 30.5, 28.8, 28.4, 28.4, 27.6, 19.6, 15.1, 12.2; HR-MS (EI) Calcd for C30H51NO6 m/z [M+]: 521.3716. Found 522.3797.
2-((2S,4aS,4bS,7S,8S,8aR,10aS)-8-(4-((tert-Butoxycarbonyl)amino)butyl)-7-(methoxycarbonyl)-4a,7-dimethyltetradecahydrophenanthren-2-yl)acetic AcidTo a solution of 13 (400 mg, 0.77 mmol) and Bu4NHSO4 (40 mg) in 1 : 1 MeOH/THF (15.4 mL) was added 10% LiOH (3.6 mL, 15.2 mmol) and stirred at r.t. for 3.5 h. The mixture was concentrated in vacuo and dilute with CH2Cl2. The mixture was neutralized with 10% KHSO4 and the aqueous layer was extracted with CH2Cl2. The concentrated residue was purified by column chromatography (CH2Cl2–MeOH=10 : 1) to give a desired carboxylic acid (389 mg, quant.) as a white amorphous.
1H-NMR (CDCl3, 300 MHz) δ: 4.51 (br, 1H), 3.64 (s, 3H), 3.04 (dd, 2H, J=11.6, 6.2 Hz), 2.22 (d, 2H, J=6.9 Hz), 1.95–0.65 (m, 25H), 1.43 (s, 9H), 1.11 (s, 3H), 0.72 (s, 3H); 13C-NMR (CDCl3, 100 MHz) δ: 179.0, 178.2, 155.7, 79.0, 53.4, 51.6, 47.9, 47.6, 46.0, 41.8, 40.4, 38.2, 37.9, 37.0, 36.0, 35.1, 35.0, 32.0, 31.8, 30.7, 29.0, 28.5, 28.5, 27.8, 19.8, 15.3, 12.4.
2-((2S,4aS,4bS,7S,8S,8aR,10aS)-8-(4-Guanidinobutyl)-7-(methoxycarbonyl)-4a,7-dimethyltetradecahydrophenanthren-2-yl)acetic Acid (3a)To a solution of the above carboxylic acid (350 mg, 0.71 mmol) in CH2Cl2 (7.1 mL) was added trifluoroacetic acid (TFA) (5.5 mL, d 1.48, 71 mmol) at 0°C, and the mixture was stirred at r.t. for 4.5 h. The reaction mixture was concentrated, and then pyridine (7.1 mL) and N,N′-bis(tert-butoxycarbonyl)-1H-pyrazole-1-carboxamidine (1.1 g, 3.55 mmol) was added to the residue. The mixture was stirred for 11 d and the reaction mixture was diluted with Et2O. The mixture was neutralized with 10% KHSO4 and the water layer was extracted by Et2O. The organic layer was dried over MgSO4 and concentrated in vacuo. The residue was purified by column chromatography (CH2Cl2–MeOH=20 : 1) to give a N-Boc guanidine derivative (462 mg, 2 steps quant.) as white amorphous. To the solution of above compound (50 mg, 0.08 mmol) in CH2Cl2 (769 µL) was added TFA (592 µL, d 1.48, 7.69 mmol) at 0°C and stirred at r.t. for 5 h. The reaction mixture was concentrated to give crude guanidine 3a (30 mg, 87%, 68% purity) as a colorless oil.
[α]D28 −5.75 (c=0.72, EtOH); IR (KBr) cm−1: 3354, 3171, 2937, 2857, 1705, 1662, 1451, 1383, 1243, 1148, 1120; 1H-NMR (CD3OD, 50°C, 500 MHz) δ: 3.64 (s, 3H), 3.11 (t, 2H, J=7.0 Hz), 2.15 (d, 2H, J=7.0 Hz), 1.92 (ddd, 1H, J=12.5, 6.7, 3.4 Hz), 1.85–0.60 (m, 24 H), 1.10 (s, 3H), 0.77 (s, 3H); 13C-NMR (CD3OD, 50°C, 100 MHz) δ: 180.5, 176.5, 158.5, 55.1, 52.1, 49.0, 47.5, 42.8, 42.4, 39.5, 39.2, 39.1, 38.3, 37.1, 36.5, 36.3, 33.4, 32.8, 30.5, 30.2, 29.6, 28.7, 20.9, 15.7, 12.7; HR-MS (FAB) Calcd for C25H44N3O4 m/z [MH+]: 450.3332. Found 450.3339.
Methyl (1S,2S,4aS,4bS,7S,8aS,10aR)-7-(2-Methoxy-2-oxoethyl)-1-(3-methoxyallyl)-2,4b-dimethyltetradecahydrophenanthrene-2-carboxylateTo a suspension of (methoxymethyl)triphenylphosphonium chloride (263 mg, 0.78 mmol) in THF (2.6 mL) was added LHMDS (765 µL, 0.765 mmol) and stirred at −78°C for 15 min under a nitrogen atmosphere. After the solution of 11 (100 mg, 0.255 mmol) in THF (2.6 mL) was added to the reaction mixture and stirred for 4.5 h. The reaction was quenched with satd aqueous NH4Cl and extracted with Et2O. The organic layer was washed with brine and dried over MgSO4. The concentrated residue was purified by column chromatography (AcOEt–n-hexane=1 : 10) to give a desired enol ether (78.4 mg, 73%) as a colorless oil.
1H-NMR (CDCl3, 300 MHz, E : Z=1 : 1.2) δ: 6.16 (d, 0.46H, J=12.6 Hz), 5.74 (dt, 0.54H, J=6.2, 1.6 Hz), 4.64 (dt, 0.46H, J=12.6, 7.6 Hz), 4.28 (dd, 0.54H, J=13.6, 7.1 Hz), 3.66 (s, 3H), 3.631 (s, 1.4H), 3.625 (s, 1.6H), 3.55 (s, 1.6H), 3.47 (s, 1.4H), 2.20 (d, 2H, J=6.9 Hz), 2.14–0.52 (m, 21H), 1.11 (s, 1.6H), 1.10 (s, 1.4H), 0.73 (s, 3H); 13C-NMR (CDCl3, 100 MHz, E : Z=1 : 1.2) δ: 179.1 (Z), 179.0 (E), 173.23 (Z), 173.21 (E), 147.0 (E), 145.0 (Z), 106.9 (Z), 102.8 (E), 59.4, 55.9, 53.5, 51.5 (Z), 51.4 (E), 48.0 (E), 47.5 (Z), 47.1, 45.9 (0.54), 45.8 (E), 41.8, 38.2, 37.9 (E), 37.7 (Z), 37.4 (0.54), 37.3 (E), 36.0, 35.21 (Z), 35.20 (E), 35.16 (Z), 35.12 (E), 31.8 (E), 31.5 (0.54), 29.2 (E), 28.93 (Z), 28.91 (E), 28.6 (Z), 25.8, 19.93 (E), 19.90 (Z), 15.4 (E), 15.3 (Z), 12.3; HR-MS (FAB) Calcd for C25H41O5 m/z [MH+]: 421.2954. Found 421.2963.
Methyl (1S,2S,4aS,4bS,7S,8aS,10aR)-7-(2-Methoxy-2-oxoethyl)-2,4b-dimethyl-1-(3-oxopropyl)tetradecahydrophenanthrene-2-carboxylate (14)To a solution of the enol ether (114 mg, 0.271 mmol) in acetone (9 mL)–H2O (300 µL) was added TsOH·H2O (262 mg, 1.35 mmol) at 0°C and stirred at r.t. for overnight. The reaction mixture was neutralized with satd aqueous NaHCO3 and extracted with Et2O. The combined organic layer was washed with brine and dried over MgSO4. The concentrated residue was purified by column chromatography (AcOEt–n-hexane=1 : 5) to give compound 14 (80.0 mg, 73%) as a colorless oil.
IR (CHCl3) cm−1: 3516, 2949, 2922, 2727, 1732, 1711, 1385, 1238; 1H-NMR (CDCl3, 300 MHz) δ: 9.62 (t, 1H, J=1.4 Hz), 3.62 (s, 3H), 3.62 (s, 3H), 2.43 (td, 2H, J=7.8, 1.4 Hz), 2.16 (d, 2H, J=7.2 Hz), 1.82–0.73 (m, 21H), 1.08 (s, 3H), 0.70 (s, 3H); 13C-NMR (CDCl3, 100 MHz) δ: 201.8, 178.6, 173.1, 53.3, 51.7, 51.3, 47.8, 46.7, 45.8, 45.0, 41.7, 38.1, 37.9, 37.2, 35.9, 35.1, 35.0, 31.9, 28.8, 28.5, 24.0, 19.7, 15.2, 12.3; HR-MS (EI) Calcd for C24H38O5 m/z [M+]: 406.2719. Found 406.2717.
Methyl (1S,2S,4aS,4bS,7S,8aS,10aR)-1-(3-((tert-Butoxycarbonyl)amino)propyl)-7-(2-methoxy-2-oxoethyl)-2,4b-dimethyltetradecahydrophenanthrene-2-carboxylate (15)To a solution of 14 (355 mg, 0.87 mmol) in EtOH (9 mL) was added Et3N (487 µL, d 0.73, 3.49 mmol) and hydroxylamine hydrochloride (152 mg, 2.18 mmol) and stirred vigorously at r.t. for 2.5 h. The reaction mixture was diluted with Et2O and washed with water and brine. The organic layer was dried over MgSO4 and concentrated in vacuo to give crude oxime. Under a hydrogen atmosphere (30 atm), a suspension of the crude oxime (165 mg, 0.39 mmol), PtO2 (16.5 mg) in AcOH (15 mL) was vigorously stirred at r.t. for 10 h in an autoclave. The reaction mixture was filtered through a Celite pad and concentrated. The residue was dissolved in CH2Cl2 (4.0 mL) and added Et3N (163 µL, d 0.73, 1.17 mmol) and Boc2O (179 µL, d 0.95, 0.78 mmol). After 2 h stirring, the reaction mixture was washed with H2O and dried over MgSO4. The concentrated residue was purified by column chromatography (AcOEt–n-hexane=1 : 5) to give compound 15 (155 mg, 78%) as a white amorphous.
[α]D27 +18.2 (c=0.51, CHCl3); IR (CHCl3) cm−1: 3455, 2947, 2924, 1715, 1506; 1H-NMR (CDCl3, 500 MHz) δ: 4.66 (br, 1H), 3.66 (s, 3H), 3.66 (s, 3H), 2.99 (quintet, 2H, J=6.2 Hz), 2.20 (d, 2H, J=7.4 Hz), 1.88–0.73 (m, 23H), 1.43 (s, 9H), 1.08 (s, 3H), 0.73 (s, 3H); 13C-NMR (CDCl3, 125 MHz) δ: 179.0, 173.3, 155.8, 78.6, 53.2, 51.4, 51.1, 47.7, 47.0, 45.7, 41.6, 40.7, 37.9, 37.7, 36.8, 35.7, 34.94, 34.89, 31.7, 30.6, 29.0, 28.7, 28.3, 19.5, 15.0, 12.1; HR-MS (FAB) Calcd for C29H50NO6 m/z [MH+]: 508.3638. Found 508.3637.
2-((2S,4aS,4bS,7S,8S,8aR,10aS)-8-(3-((tert-Butoxycarbonyl)amino)propyl)-7-(methoxycarbonyl)-4a,7-dimethyltetradecahydrophenanthren-2-yl)acetic AcidTo a solution of 15 (126 mg, 0.25 mmol) and Bu4NHSO4 (13 mg) in 1 : 1 MeOH/THF (5.0 mL) was added 10% aqueous LiOH (1.2 mL, 5.0 mmol) and stirred at r.t. overnight. The reaction mixture was concentrated in vacuo and diluted with CH2Cl2. The solution was neutralized with 10% aqueous KHSO4 at 0°C, and the aqueous layer was extracted with CH2Cl2. The concentrated residue was purified by column chromatography (CH2Cl2–MeOH=20 : 1) to give a desired carboxylic acid (122 mg, quant) as a white amorphous.
mp 176–180°C (AcOEt–n-hexane); [α]D27 +83.1 (c=0.045, CHCl3); IR (CHCl3) cm−1: 3455, 2945, 2924, 2857, 1709, 1506; 1H-NMR (CDCl3, 400 MHz) δ: 4.56 (br, 1H), 3.66 (s, 3H), 3.00 (br, 2H), 2.22 (d, 2H, J=5.1 Hz), 1.87–0.68 (m, 23H), 1.43 (s, 9H), 1.08 (s, 3H), 0.73 (s, 3H); 13C-NMR (CDCl3, 100 MHz) δ: 179.0, 178.1, 155.7, 79.0, 53.4, 51.7, 47.9, 47.2, 45.9, 41.8, 41.0, 38.2, 37.9, 37.1, 36.0, 35.1, 35.0, 32.0, 31.5, 30.9, 29.3, 28.9, 28.5, 19.8, 15.3, 12.4; HR-MS (FAB) Calcd for C28H48NO6 m/z [MH+]: 494.3482. Found: 494.3477.
2-((2S,4aS,4bS,7S,8S,8aR,10aS)-8-(3-((Z)-2,3-Bis(tert-butoxycarbonyl)guanidino)-propyl)-7-(methoxycarbonyl)-4a,7-dimethyltetradecahydrophenanthren-2-yl)acetic Acid (16)To a solution of the carboxylic acid (110 mg, 0.22 mmol) in CH2Cl2 (2.2 mL) was added TFA (1.7 mL, 22 mmol) at 0°C and the mixture was stirred at r.t. for 4.5 h. The reaction mixture was concentrated, and then pyridine (2.2 mL) and N,N′-bis(tert-butoxycarbonyl)-1H-pyrazole-1-carboxamidine (140 mg, 0.44 mmol) was added to the residue. The solution was stirred for 6 d and the reaction mixture was diluted with Et2O. The mixture was neutralized with 10% aqueous KHSO4, and the aqueous layer was extracted by Et2O. The organic layer was dried over MgSO4 and concentrated in vacuo. The residue was purified by column chromatography (CH2Cl2– MeOH=20 : 1) to give compound 16 (101 mg, 71%) as white amorphous.
IR (CHCl3) cm−1: 3688, 3327, 2928, 1719, 1634, 1616, 1578; [α]D20 −13.8 (c=0.87, CHCl3); 1H-NMR (CDCl3, 400 MHz) δ: 8.24 (t, 1H, J=4.4 Hz), 3.66 (s, 3H), 3.27 (m, 2H), 2.22 (d, 2H, J=7.1 Hz), 1.83–0.72 (m, 23H), 1.48 (s, 18H), 1.02 (s, 3H), 0.72 (s, 3H); 13C-NMR (CDCl3, 100 MHz) δ: 178.8, 178.2, 163.3, 155.8, 153.0, 82.9, 79.1, 53.4, 51.7, 47.9, 47.2, 46.0, 41.8, 41.4, 38.1, 37.8, 37.1, 36.0, 35.1, 35.0, 32.0, 30.0, 29.4, 28.9, 28.5, 28.4, 28.1, 19.8, 15.3, 12.4; HR-MS (FAB) Calcd for C34H58N3O8 m/z [MH+]: 635.4146. Found 636.4224;
2-((2S,4aS,4bS,7S,8S,8aR,10aS)-8-(3-Guanidinopropyl)-7-(methoxycarbonyl)-4a,7-dimethyltetradecahydrophenanthren-2-yl)acetic Acid (3b)To a solution of 16 (50 mg, 0.08 mmol) in CH2Cl2 (786 µL) was added TFA (606 µL, d 1.48, 7.86 mmol) at 0°C, and the mixture was stirred at r.t. for 5 h. The reaction mixture was concentrated to give a crude guanidine 3b (34 mg, quant, 69% purity) as a colorless oil.
IR (CHCl3) cm−1: 2952, 2924, 1710, 1678, 1199; [α]D27 −56.0 (c=0.03, EtOH); 1H-NMR (CDCl3, 400 MHz) δ: 3.65 (s, 3H), 3.07 (t, 2H, J=6.7 Hz), 2.17 (d, 2H, J=7.1 Hz), 1.94 (dd, 1H, J=12.5, 2.8 Hz), 1.84–0.60 (m, 22 H), 1.11 (s, 3H), 0.78 (s, 3H); 13C-NMR (CD3OD, 100 MHz) δ: 180.4, 176.6, 158.3, 55.0, 52.3, 49.1, 48.6, 47.4, 42.75, 42.69, 39.4, 39.2, 38.4, 37.1, 36.5, 36.2, 33.3, 31.0, 30.3, 30.1, 29.5, 20.8, 16.0, 12.8; HR-MS (FAB) Calcd for C24H42N3O4 m/z [M+H+]: 436.3186. Found 436.3181.
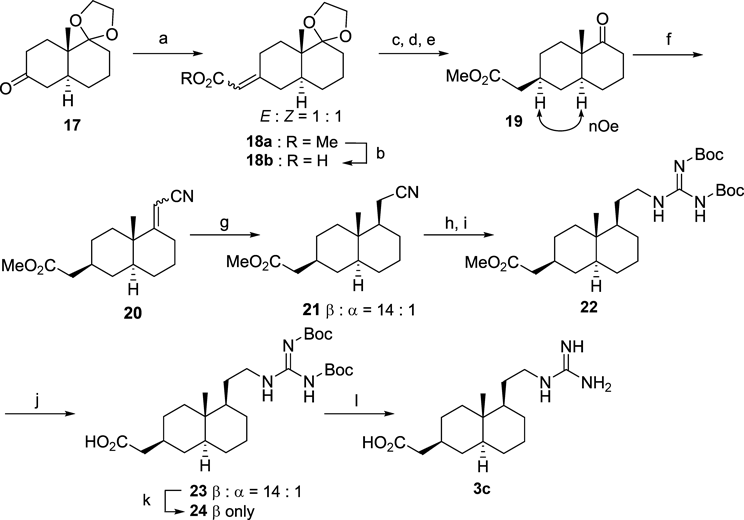
Methyl 2-((4aS,8aS)-8a-Methylhexahydro-2H-spiro[naphthalene-1,2′-[1,3]dioxolan]-6(5H)-ylidene)acetate (18a)Under a nitrogen atmosphere, to a suspension of t-BuOK (3.0 g, 27 mmol) in anhydrous DMF (22 mL) was added methyl diethylphosphonoacetate (4.9 mL, d 1.15, 27 mmol) at r.t. and stirred for 1.5 h. The mixture was cooled to 0°C and the solution of 17 (2.0 g, 8.9 mmol) in anhydrous DMF (23 mL) was added, and the resulting mixture was stirred for 12.5 h. The reaction mixture was poured into water and extracted with Et2O. The combined extracts was washed with brine, dried over MgSO4 and concentrated under the reduced pressure. The residue was purified by column chromatography (AcOEt–n-hexane=1 : 4) to give compound 18a (2.6 g, 98%) as a colorless oil.
IR (CHCl3) cm−1: 2949, 1707, 1165; [α]D26 +0.88 (c=0.90, EtOH); 1H-NMR (CDCl3, 400 MHz, E : Z=1 : 1) δ: 5.59 (1/2H, s), 5.58 (1/2H, s), 3.95-3.81 (4H, m), 3.77 (1/2H, d, J=14.4 Hz), 3.67 (3/2H, s), 3.66 (3/2H, s), 3.56 (1/2H, d, J=13.6 Hz), 2.43–1.21 (12H, m), 1.10 (3H, s); 13C-NMR (CDCl3, 100 MHz, E : Z=1 : 1) δ: 167.2, 167.1, 163.0, 162.9, 112.7, 112.54, 112.50, 112.4, 65.1, 65.0, 64.93, 64.87, 50.67, 50.66, 42.8, 42.3, 42.2, 42.0, 39.6, 33.1, 31.6, 31.4, 31.0, 30.5, 30.4, 27.9, 27.7, 24.8, 22.80, 22.77, 13.5, 13.3; HR-MS (EI) Calcd for C16H24O4 m/z [M+]: 280.1675. Found 280.1672.
2-((4aS,8aS)-8a-Methylhexahydro-2H-spiro[naphthalene-1,2′-[1,3]dioxolan]-6(5H)-ylidene)acetic Acid (18b)To a suspension of 18a (2.5 g, 8.6 mmol) in 1 : 1 EtOH/H2O (86 mL) was added KOH (4.8 g, 86 mmol), and the mixture was heated at 90°C for 0.5 h. The reaction mixture was concentrated and neutralized with 3 M HCl on an ice bath. The mixture was extracted with CH2Cl2, and the combined organic layer was dried over MgSO4. The concentrated residue was purified by column chromatography (CH2Cl2–MeOH=20 : 1) to give α,β-unsaturated carboxylic acid 18b (2.1 g, 93%) as a white amorphous.
IR (CHCl3) cm−1: 2933, 1685, 1631; [α]D26 −12.7 (c=0.06, EtOH); 1H-NMR (CDCl3, 400 MHz, E : Z=1 : 1) δ: 11.8 (1H, br s), 5.61 (1/2H, s), 5.60 (1/2H, s), 3.98–3.82 (4H, m), 3.77 (1/2H, d, J=14.8 Hz), 3.55 (1/2H, d, J=13.2 Hz), 2.35 (1/2H, dt, J=4.8, 13.6 Hz), 2.35–2.10 (m, 1H), 2.05–1.90 (m, 1H), 1.87–1.45 (m, 15/2H), 1.41–1.17 (m, 2H), 1.10 (s, 3/2H), 1.09 (s, 3/2H); 13C-NMR (CDCl3, 100 MHz, E : Z=1 : 1) δ: 172.3, 172.2, 165.8, 165.6, 112.8, 112.6 (2C), 112.5, 65.14, 65.08, 65.0, 64.9, 42.9, 42.3, 42.2, 42.1, 39.9, 33.3, 31.6 (2C), 31.1, 30.5, 30.4, 27.9, 27.7, 25.1, 22.80, 22.77, 13.5, 13.4; HR-MS (EI) Calcd for C15H22O4 m/z [M+]: 266.1518. Found 266.1517.
Methyl 2-((2S,4aS,8aS)-4a-Methyl-5-oxodecahydronaphthalen-2-yl)acetate (19)Under a nitrogen atmosphere, a lithium wire (0.5 g, 68 mmol) was added to liquid ammonia (96 mL) at −78°C. To this mixture was added a solution of 18b (1.8 g, 6.8 mmol) in THF (45 mL), and the resulting mixture was stirred for 3 h. After the substrate disappearance was monitoring by TLC, solid NH4Cl was added until the mixture turned white suspension. The mixture was allowed to r.t. and the liquid ammonia was evaporated through the night. The residue was suspended with CH2Cl2 and filtrated. To the concentrated residue in THF (68 mL) was added 1.5 M HCl (0.11 L) using a dropping funnel at 0°C, and the mixture and stirred for 1 h at r.t. The reaction mixture was extracted with CH2Cl2 and dried over MgSO4. To the concentrated residue was added MeI (6.9 mL, 68 mmol) and K2CO3 (1.9 g, 14 mmol), and then the mixture was stirred at r.t. overnight. The reaction was concentrated, diluted with Et2O and washed with H2O. The organic layer was dried over MgSO4 and concentrated. The crude material was purified by column chromatography (AcOEt–n-hexane=1 : 2) to give compound 19 (1.5 g, 91%, in 3 steps) as a colorless oil.
IR (CHCl3) cm−1: 3010, 2933, 2862, 1730, 1699; [α]D20 −7.9 (c=0.15, EtOH); 1H-NMR (CDCl3, 300 MHz) δ: 3.67 (3H, s), 2.66–2.63 (1H, m), 2.28–2.17 (3H, m), 2.08–1.96 (1H, m), 1.84–1.41 (9H, m), 1.26–1.12 (2H, m), 1.09 (3H, s); 13C-NMR (CDCl3, 100 MHz) δ: 215.9, 173.1, 51.3, 47.8, 45.2, 41.3, 37.3, 34.6, 33.8, 32.1, 27.7, 27.3, 26.0, 15.5; HR-MS (EI) Calcd for C14H22O3 m/z [M+]: 238.1569. Found 238.1562.
Methyl 2-((2S,4aS,8aS)-5-(Cyanomethylene)-4a-methyldecahydronaphthalen-2-yl)acetate (20)To a suspension of KOtBu (3.6 g, 32 mmol) in DMF (30 mL) was added diethyl cyanomethylphosphonate (5.0 mL, d 1.13, 32 mmol) under a nitrogen atmosphere, and the mixture was stirred at 0°C for 1 h. To the solution was added 19 (2.5 g, 11 mmol) in anhydrous DMF (25 mL) and the resulting mixture was stirred at 90°C for 1 h. The reaction mixture was added H2O and extracted with Et2O. The combined extracts was dried over MgSO4 and concentrated. The residue was purified by column chromatography (AcOEt–n-hexane=1 : 5) to give compound 20 (2.6 g, 95%) as a colorless oil.
IR (CHCl3) cm−1: 2983, 2929, 2216, 1730; [α]D25 −71.9 (c=0.46, EtOH); 1H-NMR (CDCl3, 400 MHz) δ: 5.03 (1H, d, J=1.6 Hz), 3.67 (3H, s), 2.94–2.78 (1H, m), 2.42–2.30 (1H, m), 2.29–2.10 (2H, m), 1.98–1.67 (3H, m), 1.66–1.57 (1H, m), 1.51–1.03 (8H, m), 1.01 (3H, s); 13C-NMR (CDCl3, 100 MHz) δ: 175.9, 173.0, 117.7, 90.2, 51.3, 45.2, 41.2, 40.6, 35.5, 34.58, 34.55, 30.1, 28.2, 28.0, 27.0, 16.9; HR-MS (EI) Calcd for C16H23NO2 m/z [M+]: 261.1729. Found 261.1725.
Methyl 2-((2S,4aS,5R,8aS)-5-(Cyanomethyl)-4a-methyldecahydronaphthalen-2-yl)acetate (21)Under a hydrogen balloon atmosphere, a suspension of compound 20 (12 mg, 0.046 mmol), 10% Pd/C (2.0 mg) in EtOH (1.4 mL) was stirred at r.t. for 1.5 h. The reaction mixture was filtered through a Celite pad and concentrated. The residue was purified by column chromatography (AcOEt–n-hexane=1 : 5) to give compound 21 (12 mg, quant, β : α=14 : 1) as a colorless oil.
IR (CHCl3) cm−1: 2927, 2858, 2247, 1730; [α]D23 +100 (c=0.004, EtOH); 1H-NMR (CDCl3, 400 MHz) δ: 3.66 (3H, s), 2.63–2.35 (1H, m), 2.21 (2H, d, J=7.2 Hz), 2.00 (1H, dd, J=16.4, 10.4 Hz), 1.90–1.71 (3H, m), 1.67–1.56 (2H, m), 1.53–0.96 (10H, m), 0.72 (3H, s); 13C-NMR (CDCl3, 100 MHz) δ: 173.3, 120.1, 51.4, 45.7, 45.6, 41.5, 38.1, 36.3, 35.0, 34.8, 28.5, 28.2, 27.8, 25.9, 18.3, 10.9; HR-MS (EI) Calcd for C16H25NO2 m/z [M+]: 263.1885. Found 263.1881.
Methyl 2-((2S,4aS,5R,8aS)-5-(2-((Z)-2,3-Bis(tert-butoxycarbonyl)guanidino)ethyl)-4a-methyldecahydronaphthalen-2-yl)acetate (22)Under a hydrogen atmosphere (balloon), a suspension of compound 21 (0.11 g, 0.45 mmol) and PtO2 (20 mg) in 10% HCl–MeOH (12 mL) was stirred at r.t. for 140 min. The reaction mixture was filtered through a Celite pad and concentrated. The residue was dissolved in DMF (3.9 mL), and Et3N (0.16 mL, d 0.73, 1.2 mmol) and N,N′-bis(tert-butoxycarbonyl)-1H-pyrazole-1-carboxamidine (0.15 g, 0.47 mmol) was added. After 1 h stirring, the reaction mixture was added H2O, extracted with Et2O, and dried over MgSO4. The concentrated residue was purified by column chromatography (AcOEt–n-hexane=1 : 5) to give compound 22 (114 mg, 83%, 2 steps) as a colorless amorphous.
IR (CHCl3) cm−1: 2981, 2926, 2856, 1720, 1633; [α]D26 +2.2 (c=0.09, EtOH); 1H-NMR (CDCl3, 400 MHz) δ: 11.5 (1H, br s), 8.25 (1H, br s), 3.66 (3H, s), 3.62–3.22 (2H, m), 2.20 (2H, d, J=6.8 Hz), 1.99–1.38 (10H, m), 1.26 (9H, s), 1.22–0.70 (13H, m), 0.53 (3H, s); 13C-NMR (CDCl3, 100 MHz) δ: 173.4, 163.6, 156.0, 153.3, 82.9, 79.1, 51.3, 46.3, 45.9, 41.7, 40.2, 37.9, 36.3, 35.2, 29.6, 28.9, 28.8, 28.4, 28.3, 28.0, 27.7, 26.3, 11.1; HR-MS (FAB) Calcd for C27H47N3O6 m/z [M+]: 510.3543. Found 510.3543.
2-((2S,4aS,5R,8aS)-5-(2-((Z)-2,3-Bis(tert-butoxycarbonyl)guanidino)ethyl)-4a-methyldecahydronaphthalen-2-yl)acetic Acid (24)To a solution of 22 (1.5 g, 3.1 mmol, β : α=14 : 1) in MeOH–THF (62 mL, 1 : 1) was added 10% aqueous LiOH (15 mL, 0.2 M) and stirred at r.t. for 4 h. The mixture was concentrated in vacuo and diluted with CH2Cl2. The solution was neutralized with dil HCl and aqueous layer was extracted with CH2Cl2 and dried over MgSO4. The concentrated residue was purified by column chromatography (CH2Cl2– MeOH=10 : 1) to give compound 23 (1.2 g, 75%, β : α=14 : 1) as a white solid. This product was recrystallized from Et2O to give β-isomer 24 as a white powder (0.20 g, 13%).
mp 108–112°C (AcOEt–n-hexane); IR (CHCl3) cm−1: 3332, 2926, 1720, 1641, 1394; [α]D21 −6.6 (c=0.09, EtOH); 1H-NMR (CDCl3, 400 MHz) δ: 8.24 (1H, brs), 3.49–3.38 (1H, m), 3.33–3.24 (1H, m), 2.24 (2H, d, J=6.8 Hz), 1.89–1.58 (8H, m), 1.50 (18H, s), 1.43–0.92 (11H, m), 0.69 (3H, s); 13C-NMR (CDCl3, 100 MHz) δ: 178.7, 163.5, 156.0, 153.7, 83.0, 79.2, 46.3, 41.7, 40.3, 37.9, 36.3, 36.1, 35.1, 28.89, 28.85, 28.4, 28.3, 28.0, 27.6, 26.3, 11.1; HR-MS (FAB) Calcd for C26H46N3O6 m/z [MH+]: 496.3387. Found 496.3384.
2-((2S,4aS,5R,8aS)-5-(2-Guanidinoethyl)-4a-methyldecahydronaphthalen-2-yl)acetic Acid (3c)To a solution of 24 (39 mg, 0.080 mmol) was added HCl–AcOEt (0.80 mL, 0.4 M) at 0°C and stirred at r.t. for 31 h. The mixture was concentrated in vacuo to give compound 3c (24 mg, quant, 70% purity) as a white amorphous.
IR (CHCl3) cm−1: 3435, 3417, 1633, 1261, 1095, 1022, 802; [α]D23 +22.7 (c=0.22, EtOH); 1H-NMR (CDCl3, 300 MHz) δ: 7.48–7.35 (1H, m), 3.24–2.82 (2H, m), 2.10 (2H, d, J=7.2 Hz), 1.90–0.83 (20H, m), 0.66 (3H, s); 13C-NMR (CDCl3, 125 MHz) δ: 173.6, 156.6, 53.5, 52.3, 49.4, 37.5, 35.9, 34.7, 28.5, 28.3, 27.8, 27.6, 26.9, 25.9, 11.0, 0.05; HR-MS (FAB) Calcd for C16H30N3O2 m/z [MH+]: 296.2338. Found 296.2335.
(3aS,7aS)-7a-Methylhexahydrospiro[indene-1,2′-[1,3]dioxolan]-5(4H)-one (25)Under a nitrogen atmosphere, a lithium wire (0.16 g, 23.0 mmol) was added to liquid ammonia (30 mL) at −78°C. To the reaction mixture was added the solution of acetal (1.5 g, 7.2 mmol) in THF (18 mL), and the resulting mixture was stirred for 100 min. After the substrate disappearance was monitored by TLC, solid NH4Cl was added until the mixture turned white suspension. The mixture was allowed to r.t. and the liquid ammonia was evaporated through the night. The residue was suspended with CH2Cl2 and filtrated. The concentrated residue was purified by column chromatography (AcOEt–n-hexane=1 : 4) to give compound 25 (1.0 g, 70%) as a colorless oil.
IR (CHCl3) cm−1: 2956, 1705; [α]D22 +58.8 (c=0.17, EtOH); 1H-NMR (CDCl3, 400 MHz) δ: 3.97–3.87 (4H, m), 2.52–2.19 (5H, m), 2.02–1.78 (4H, m), 1.72–1.66 (1H, m), 1.42–1.30 (1H, m), 1.16 (3H, s); 13C-NMR (CDCl3, 100 MHz) δ: 213.0, 119.4, 65.2, 64.4, 44.3, 44.1, 42.2, 37.1, 32.9, 29.4, 25.7, 18.4; HR-MS (EI) Calcd for C12H18O3 m/z [M+]: 210.1256. Found 210.1257.
Methyl 2-((3aS,7aS)-7a-Methylhexahydrospiro[indene-1,2′-[1,3]dioxolan]-5(4H)-ylidene)acetateTo a suspension of KOtBu (0.11 g, 1.1 mmol) in DMF (0.75 mL) was added methyl 2-(diethoxyphosphoryl)acetate (0.19 mL, d 1.15, 1.1 mmol), and the mixture was stirred at 0°C for 10 min under a nitrogen atmosphere. To the mixture was added compound 25 (74 mg, 0.35 mmol) in anhydrous DMF (1.0 mL) and the resulting mixture was stirred at r.t. for 30 h. The reaction mixture was added H2O and extracted with Et2O. The combined extract was dried over MgSO4 and concentrated. The residue was purified by column chromatography (AcOEt–n-hexane=1 : 4) to give α,β-unsaturated ester (90 mg, 97%) as a colorless oil.
IR (CHCl3) cm−1: 3012, 2949, 2879, 1708, 1649; [α]D21 +65.5 (c=0.07, EtOH); 1H-NMR (CDCl3, 400 MHz, E : Z=1 : 1) δ: 5.76 (1/2H, s), 5.64 (1/2H, s), 3.95–3.81 (4H, m), 3.69 (3/2H, s), 3.68 (3/2H, s), 3.62–3.50 (1H, m), 2.47–1.19 (10H, m), 1.06 (3/2H, s), 1.05 (3/2H, s); 13C-NMR (CDCl3, 100 MHz, E : Z=1 : 1) δ: 167.0, 166.7, 161.7, 161.6, 120.0 (2C), 115.0, 114.6, 65.2 (2C), 64.2, 64.1, 50.7 (2C), 44.7, 44.6, 43.9, 43.8, 37.2, 33.1, 32.7, 32.6, 31.3, 29.8, 28.7, 25.1, 23.9, 23.5, 17.6, 16.9; HR-MS (EI) Calcd for C15H22O4 m/z [M+]: 266.1518. Found 266.1516.
2-((3aS,7aS)-7a-Methylhexahydrospiro[indene-1,2′-[1,3]dioxolan]-5(4H)-ylidene)acetic Acid (26)To a solution of α,β-unsaturated ester (57 mg, 0.21 mmol) in 1 : 1 EtOH/H2O (2.0 mL) was added powdered KOH (0.12 g, 2.1 mmol) and the mixture was heated at 90°C for 15 min. The reaction mixture was concentrated and neutralized with 10% HCl on an ice bath. The mixture was extracted with CH2Cl2 and the combined organic layer was dried over MgSO4. The concentrated residue was purified by column chromatography (AcOEt–n-hexane=1 : 3) to give compound 26 (38 mg, 71%) as a white amorphous.
IR (CHCl3) cm−1: 3016, 2949, 2879, 1687; [α]D25 +100 (c=0.02, EtOH); 1H-NMR (CDCl3, 400 MHz, E : Z=1 : 1) δ: 5.78 (1/2H, s), 5.67 (1/2H, s), 3.95–3.79 (4H, m), 3.55 (1H, d, J=16.0 Hz), 2.49–1.10 (11H, m), 1.06 (3/2H, s), 1.05 (3/2H, s); 13C-NMR (CDCl3, 100 MHz, E : Z=1 : 1) δ: 165.6, 165.5, 164.8, 164.6, 120.1, 120.1, 114.8, 114.3, 65.3, 64.3, 64.2, 44.7, 44.7, 44.1, 44.0, 37.6, 33.5, 32.8, 32.7, 31.3, 29.8, 29.1, 25.4, 24.1, 23.7, 17.9, 17.0; HR-MS (EI) Calcd for C14H20O4 m/z [M+]: 252.1362. Found 252.1356.
Methyl 2-((3aS,5S,7aS)-7a-Methyl-1-oxooctahydro-1H-inden-5-yl)acetate (27)Under a nitrogen atmosphere, a lithium wire (0.74 g, 0.10 mol) was added to liquid ammonia (150 mL) at −78°C. To the reaction mixture was added the solution of 26 (2.7 g, 11 mmol) in THF (100 mL), and the mixture was stirred for 25 min at r.t. After the substrate disappearance was monitored by TLC, solid NH4Cl was added until the mixture turned white suspension. The mixture was allowed to r.t. and the liquid ammonia was evaporated through the night. The residue was suspended with CH2Cl2 and filtrated. To the concentrated residue in THF (100 mL) was added 1.5 M HCl (40 mL) using a dropping funnel at 0°C, and the mixture and stirred for 1.5 h at r.t. The reaction mixture was extracted with CH2Cl2 and dried over MgSO4. To the concentrated residue was dissolved in acetone (100 mL), and MeI (11 mL, 0.10 mol) and K2CO3 (6.0 g, 43 mmol) were added, and then the mixture was stirred for overnight. The reaction was concentrated, diluted with Et2O and washed with H2O. The organic layer was dried over MgSO4 and concentrated. The crude material was purified by column chromatography (AcOEt–n-hexane=1 : 2) to give compound 27 (2.4 g, 98%, 3 steps) as a colorless oil.
IR (CHCl3) cm−1: 2953, 2929, 2358, 2339, 1730; [α]D20 +35.6 (c=0.03, EtOH); 1H-NMR (CDCl3, 400 MHz) δ: 3.68 (3H, s), 2.54–2.38 (1H, m), 2.32–1.80 (7H, m), 1.75–1.56 (2H, m), 1.50–1.35 (2H, m), 1.29–1.10 (2H, m), 1.07 (3H, s); 13C-NMR (CDCl3, 100 MHz) δ: 222.2, 172.9, 51.2, 46.6, 42.3, 41.0, 35.9, 30.9, 29.0, 27.7, 27.3, 22.9, 18.8; HR-MS (EI) Calcd for C13H20O3 m/z [M+]: 224.1412. Found 224.1412.
Methyl 2-((3aS,5S,7aS)-1-(Cyanomethylene)-7a-methyloctahydro-1H-inden-5-yl)acetateTo a suspension of NaH (26 mg, 60% oil suspension, 0.66 mmol) in DMF (0.50 mL) was added diethyl cyanomethylphosphonate (0.10 mL, d 1.13, 0.66 mmol) and stirred at 0°C for 1 h under a nitrogen atmosphere. To the solution was added compound 11 (550 mg, 0.22 mmol) in anhydrous DMF (0.51 mL) and stirred at r.t. for 11 h. The reaction mixture was added H2O and extracted with Et2O. The combined extract was dried over MgSO4 and concentrated. The residue was purified by column chromatography (Et2O–n-hexane=1 : 4) to give the desired α,β-unsaturated nitrile (50 mg, 93%) as a colorless oil.
IR (CHCl3) cm−1: 3018, 2954, 2927, 2216, 1730; [α]D26 +11.7 (c=0.29, EtOH); 1H-NMR (CDCl3, 400 MHz) δ: 5.10 (1H, t, J=2.4 Hz), 3.65 (3H, s), 2.90–2.69 (1H, m), 2.57–2.46 (1H, m), 2.21 (2H, d, J=6.8 Hz), 2.07–1.15 (10H, m), 1.07 (3H, s); 13C-NMR (CDCl3, 100 MHz) δ: 182.1, 172.9, 117.3, 89.2, 51.3, 44.9, 44.8, 41.0, 33.0, 30.9, 30.2, 28.8, 28.1, 26.0, 21.8; HR-MS (EI) Calcd for C15H21NO2 m/z [M+]: 247.1572. Found 247.1572.
Methyl 2-((1R,3aS,5S,7aS)-1-(Cyanomethyl)-7a-methyloctahydro-1H-inden-5-yl)acetate (28)Under a hydrogen balloon atmosphere, a suspension of the α,β-unsaturated nitrile (23 mg, 0.090 mmol), 10% Pd/C (2.3 mg) in EtOH (3.0 mL) was stirred at r.t. for 2 h. The reaction mixture was filtered through a Celite pad and concentrated. The residue was purified by column chromatography (AcOEt–n-hexane=1 : 5) to give nitrile 28 (18.6 mg, 80%, β: α=10 : 1) as a colorless oil.
IR (CHCl3) cm−1: 2953, 2926, 1730; [α]D26 −29.0 (c=0.06, EtOH); 1H-NMR (CDCl3, 400 MHz) δ: 3.67 (3H, s), 2.40–2.25 (1H, m), 2.20 (2H, d, J=5.6 Hz), 2.17–1.03 (14H, m), 1.02 (30/11H, s), 0.95 (3/11, s); 13C-NMR (CDCl3, 100 MHz) δ: 173.3, 119.8, 51.4, 47.9, 45.6, 41.6, 41.0, 31.0, 29.0, 27.9, 27.6, 26.8, 25.5, 22.6, 17.1; HR-MS (EI) Calcd for C15H23NO2 m/z [M+]: 249.1729. Found 249.1727.
Methyl 2-((1R,3aS,5S,7aS)-1-(2-((E)-2,3-Bis(tert-butoxycarbonyl)guanidino)ethyl)-7a-methyloctahydro-1H-inden-5-yl)acetateUnder a hydrogen atmosphere (balloon), a suspension of nitrile 28 (29 mg, 0.11 mmol) and PtO2 (3 mg) in 10% HCl–MeOH (2.7 mL) was stirred at r.t. for 2 h. The reaction mixture was filtered through a Celite pad and concentrated. The residue was dissolved in DMF (1.1 mL), and Et3N (45 µL, d 0.73, 0.33 mmol) and N,N′-bis(tert-butoxycarbonyl)-1H-pyrazole-1-carboxamidine (40 mg, 0.13 mmol) were added. After 1 h stirring, the reaction mixture was added H2O, extracted with Et2O, and dried over MgSO4. The concentrated residue was purified by column chromatography (AcOEt–n-hexane=1 : 5) to give N,N′-bisBoc guanidine (11 mg, 13%) as a colorless amorphous.
IR (CHCl3) cm−1: 2927, 1716, 1635; [α]D21 +15.0 (c=0.02, EtOH); 1H-NMR (CDCl3, 400 MHz, 10 : 1 diastereo mixture) δ: 11.5 (1H, s), 8.27 (1H, s), 3.66 (3H, s), 3.43–3.33 (2H, m), 2.25 (2/11H, d, J=7.6 Hz), 2.18 (20/11H, d, J=7.2 Hz), 2.14–1.78 (2H, m), 1.77–1.54 (5H, m), 1.51 (9H, s), 1.49 (9H, s), 1.47–0.98 (8H, m), 0.94 (30/11H, s), 0.86 (3/11H, s); 13C-NMR (CDCl3, 100 MHz) δ: 173.6, 163.6, 156.0, 153.3, 83.0, 79.2, 51.4, 49.6, 45.7, 41.8, 41.1, 40.7, 31.3, 29.3, 29.2, 28.30, 28.26, 28.1, 27.7, 27.1, 26.1, 22.7; HR-MS (FAB) Calcd for C26H46N3O6 m/z [MH+]: 496.3386. Found 496.3390.
2-((1R,3aS,5S,7aS)-1-(2-((E)-2,3-Bis(tert-butoxycarbonyl)guanidino)ethyl)-7a-methyloctahydro-1H-inden-5-yl)acetic AcidTo a solution of the N,N′-bisBoc guanidine (36 mg, 0.070 mmol) in MeOH (0.70 mL) and THF (0.70 mL) was added 10% aqueous LiOH (0.70 mL, 0.1 M), and the mixture was heated at r.t. for 4 h. The reaction mixture was concentrated and neutralized with dil HCl on an ice bath. The mixture was extracted with CH2Cl2, and the combined organic layer was dried over MgSO4. The concentrated residue was purified by column chromatography (AcOEt–n-hexane=1 : 1) to give the desired carboxylic acid (28 mg, 82%) as a white amorphous.
IR (CHCl3) cm−1: 2981, 2927, 2872, 1714, 1633; 1H-NMR (CDCl3, 400 MHz) δ: 11.5 (1H, s), 8.27 (1H, s), 3.53–3.33 (2H, m), 2.18 (2H, d, J=7.6 Hz), 2.14–1.54 (7H, m), 1.53 (9H, s), 1.49 (9H, s), 1.47–0.98 (9H, m), 0.94 (3H, s); 13C-NMR (CDCl3, 100 MHz) δ: 156.0, 153.3, 83.0, 79.3, 53.4, 49.6, 45.6, 41.1, 40.7, 31.2, 31.0, 29.1, 28.3, 28.2, 28.1, 27.7, 27.1, 26.1, 22.7; HR-MS (FAB) Calcd for C25H44N3O6 m/z [MH+]: 481.3230. Found 482.3233.
2-((1R,3aS,5S,7aS)-1-(2-Guanidinoethyl)-7a-methyloctahydro-1H-inden-5-yl)acetic Acid (3d)To a solution of the carboxylic acid (24 mg, 0.050 mmol) was added 4 M HCl–AcOEt (0.5 mL) at 0°C, and the mixture was stirred at r.t. for 6.5 h. The mixture was concentrated in vacuo to give compound 3d (7.8 mg, 56, 75% purity) as a pale yellow oil.
IR (CHCl3) cm−1: 3152, 2956, 2928, 2868, 1725, 1678, 1642, 1615, 1252, 1148; [α]D20 −5.0 (c=0.12, EtOH); 1H-NMR (DMSO-d6, 300 MHz) δ: 11.95 (5H, br s), 8.07 (1H, br s), 7.15 (3H, brs), 3.24–2.97 (2H, m), 2.08 (2H, d, J=9.2 Hz), 1.08–0.97 (15H, m), 0.92 (3H, s); 13C-NMR (DMSO-d6, 100 MHz) δ: 173.7, 171.9, 156.7, 48.5, 45.4, 41.4, 30.7, 28.6, 28.4, 27.7, 26.9, 26.6, 25.6, 22.6, 21.0; HR-MS (FAB) Calcd for C15H28N3O2 m/z [MH+]: 282.2181. Found 282.2184.
Methyl 2-((2S,4aS,5S,8aS)-5-Hydroxy-4a-methyldecahydronaphthalen-2-yl)acetate (29)To a solution of 19 (10 mg, 0.040 mmol) in MeOH (0.2 mL) was added NaBH4 at 0°C, and the mixture was stirred at r.t. for 5 min. The reaction mixture was concentrated and then purified by column chromatography (AcOEt–n-hexane=1 : 4) to give compound 29 (8.0 mg, 83%, β : α=8.5 : 1) as a colorless oil.
IR (CHCl3) cm−1: 2927, 2858, 1730; [α]D25 −13.0 (c=0.26, EtOH); 1H-NMR (CDCl3, 400 MHz) δ: 3.67 (3H, s), 3.23 (1H, dd, J=12.0, 4.0 Hz), 2.22 (2H, d, J=4.0 Hz), 2.60–0.90 (15H, m), 0.79 (3H, s); 13C-NMR (CDCl3, 100 MHz) δ: 173.5, 79.5, 51.4, 43.5, 41.7, 38.7, 36.9, 35.2, 34.2, 30.4, 28.1, 27.8, 24.3, 9.8; HR-MS (EI) Calcd for C14H24O3 m/z [M+]: 240.1725. Found 240.1724.
2-((2S,4aS,5S,8aS)-5-(3-(1-(tert-Butoxycarbonyl)piperidin-4-yl)propoxy)-4a-methyldecahydronaphthalen-2-yl)acetic Acid (30)To a solution of 29 (22 mg, 0.090 mmol, β : α=8.5 : 1) in anhydrous DMF (1.0 mL) was added NaH (5.5 mg, 0.23 mmol, 60% oil suspension), and the mixture was stirred at r.t. for 1 h under a nitrogen atmosphere. The mixture was added tert-butyl 4-(3-bromopropyl)piperidine-1-carboxylate (50 mg, 0.090 mmol) and stirred for 30 h. Then the solution was neutralized with satd aqueous NH4Cl and extracted with Et2O. The extract was washed with brine and dried over Na2SO4. The concentrated residue was purified by column chromatography (AcOEt–n-hexane=1 : 3) to give compound 30 (12 mg, 29%) as a colorless oil and 29 (12 mg, 53%) was recovered.
IR (CHCl3) cm−1: 3444, 2929, 2358, 1722, 1678; [α]D28 +3.0 (c=0.82, CHCl3); 1H-NMR (CDCl3, 500 MHz) δ: 4.05 (2H, t, J=6.5 Hz), 3.23 (1H, dd, J=4.0 Hz), 2.68 (2H, t, J=12.5 Hz), 2.20 (2H, d, J=6.0 Hz), 1.93–1.47 (11H, m), 1.45 (9H, s), 1.43–0.84 (15H, m), 0.80 (3H, s); 13C-NMR (CDCl3, 100 MHz) δ: 173.1, 154.9, 79.5, 79.2, 64.3, 43.7, 43.5, 42.0, 38.7, 36.9, 35.6, 35.3, 34.2, 32.7, 32.1, 30.4, 28.4, 28.0, 27.8, 25.8, 24.3, 24.0, 10.9, 9.8; HR-MS (EI) Calcd for C26H45NO5 m/z [M+]: 451.3298. Found 451.3295.
2-((2S,4aS,5S,8aS)-4a-Methyl-5-(3-(piperidin-4-yl)propoxy)decahydronaphthalen-2-yl)acetic Acid (3e)To a solution of 30 (6.2 mg, 0.010 mmol) was added 4 M HCl–AcOEt (0.1 mL) at 0°C, and the mixture was stirred at r.t. for 5 h. The reaction mixture was concentrated in vacuo to give compound 3e (3.6 mg, 75, 72% purity) as a white amorphous.
IR (CHCl3) cm−1: 3685, 2928, 2857, 1722, 1601, 1453; [α]D28 +2.4 (c=0.45, EtOH); 1H-NMR (CD3OD, 400 MHz) δ: 4.21–4.02 (2H, m), 3.37 (2H, d, J=12.4 Hz), 3.15 (1H, dd, J=4.8, 11.2 Hz), 2.96 (2H, t, J=12.4 Hz), 2.21 (2H, d, J=7.2 Hz), 2.12–0.90 (25H, m), 0.90 (1H, s), 0.79 (2H, s); 13C-NMR (CD3OD, 100 MHz) δ: 174.8, 80.2, 65.3, 45.3 (2C), 45.2, 42.8, 40.0, 38.4, 36.8, 35.5, 34.5, 33.4, 31.3, 30.0 (2C), 29.19, 29.16, 26.7, 25.6, 10.3; HR-MS (FAB) Calcd for C21H37NO3 m/z [MH+]: 352.2852. Found 352.2857.
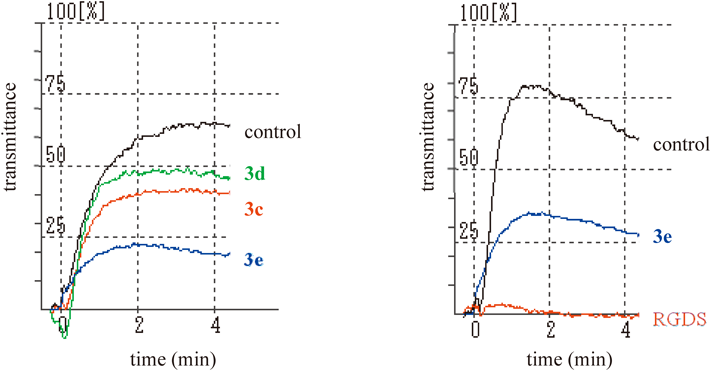
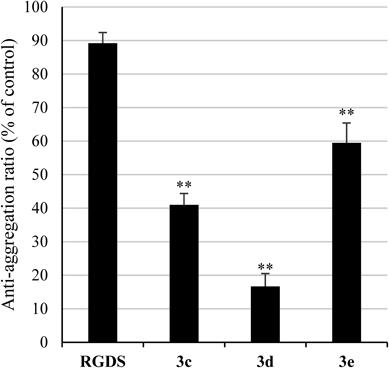
Platelet Aggregation AssayRabbit platelet rich-plasma (PRP) was obtained from fresh rabbit blood collected into a one-tenth volume of 3.8% sodium citrate by centrifugation at 1000 rpm (50×g) for 20 min at r.t. Platelet aggregometry followed the methods of Born39) and O’Brien.40) Rabbit PRP (175 µL) was added to 10 mM CaCl2 (25 µL) and the mixture was preincubated at 37°C for 1 min. The mixture was added to each sample in 0.02 M Tris–HCl buffer (pH 7.4) containing 0.15 M NaCl (25 µL) and incubated at 37°C for 5 min. A total of 25 µL of ADP (final concentration: 1–2 µM) or collagen (final concentration: 1 µg/mL) was then added and incubated at 37°C for 5–7 min. Platelet aggregation was measured by the turbidimetric method using HEMA TRACER 704 from MC MEDICAL Inc. (Tokyo, Japan).
Statistical AnalysisStatistical analyses were performed by using Microsoft Excel 2007 software (Microsoft Co., Redmond, WA, U.S.A.). The statistical significance of differences was determined according to the student’s t-test. p<0.05 was considered to be statistically significant.