Regular Articles
Physicochemical Evaluation and Developability Assessment of Co-amorphouses of Low Soluble Drugs and Comparison to the Co-crystals
2016 年 64 巻 12 号 p. 1739-1746

詳細
2016 年 64 巻 12 号 p. 1739-1746
To judge the developability and analyze functional mechanism of co-amorphouses, we investigated the physicochemical properties of co-amorphouses and compare the properties with the co-crystals having the same drug and counters. Co-amorphous compounds are a novel approach to improve the physicochemical properties of drugs. A co-amorphous is in an amorphous solid state allowing non-ionic interactions between drug molecules and counter molecules. The co-amorphous compounds composed of itraconazole (ITZ) with the organic carboxyl acid, fumaric acid (FA) or L-tartaric acid (TA), were prepared by mechanical grinding. Potential interactions within ITZ-FA co-amorphous were assessed by Raman spectroscopy. ITZ-FA co-amorphous was not crystallized as the co-crystal or as a single ITZ crystal, suggesting that the amorphous state, like the amorphous solid dispersion, was physically stable and that ITZ-FA co-amorphous was also chemically stable. In contrast, no clear interactions were observed within ITZ-TA co-amorphous, and the co-amorphous was physically stable but chemically unstable. The solubility of the co-amorphous state was much higher than those of ITZ crystal and the co-crystals and was almost identical to that of amorphous ITZ. A co-amorphous compound like ITZ-FA co-amorphous might be feasible to implement in the development of solid drug products and bring some merits compared to the co-crystals, and the function is governed by the interaction between a drug and a counter. The co-amorphous approach may be an effective strategy for drug development and can contribute to the production of novel drugs with improved functions.
Solubility improvement is one of the most important challenges for drug development of poorly soluble drugs. Amorphization of drugs is a major effective strategy to prepare solubilized formulations, because the crystal packing effect on a drug substance significantly decreases its solubility.1–3) An amorphous compound is in a higher energy state and has a higher mobility than a crystalline compound, improving its solubility and dissolution properties. However, due to the higher energy state, the physical and chemical stability of amorphous compounds are relatively lower than those of a crystal.4,5) In many cases, an amorphous drug is formulated by solid dispersion with hydrophilic polymers to improve the amorphous drug stability.6–8) The complex formed with polymers can prevent the drug’s crystallization and maintain the physical stability of the amorphous state. Generally, weak interactions cannot sufficiently stabilize amorphous drugs.9) The prediction and design leading to the interactions between the drug and the polymer are still difficult to accomplish. Therefore, solid dispersions combined with several kinds and amounts of polymers need to be tested.
As another approach to enhance a drug’s solubility, pharmaceutical co-crystals have been investigated and adapted to the drug development processes in concurrence with pharmaceutical salts.10,11) Co-crystals contain an interaction unit between a drug molecule and a small counter molecule, and, unlike for a salt, no proton transfer is observed between the drug molecule and the counter molecule. The interaction unit is called as a synthon, which is formed mainly by hydrogen bonds.12–14) Co-crystals are a kind of crystals having low energy state then show relatively better stability than amorphous state. However, some co-crystals do not always have higher solubility than the free form of drugs due to the interaction strength with counter molecules and the crystal lattice energy. And, crystallization of co-crystals, especially in a large-scale process, can be difficult because of the solubility difference between a drug and the counter molecules.15)
In recent years, co-amorphouses heve been investigated actively. Co-amorphouses are amorphous complexes with specific interactions, which are seemed to be synthon, between drugs and counters or other drugs, i.e., have both features of solid dispersion and co-crystal.16–19) Thus, co-amorphouses have been studied for their improvement of the physicochemical properties of drugs, especially the solubility.20,21) In certain cases, a co-amorphous was physically more stable because of the presence of a synthon compared with the stability of each single, non-interacting amorphous molecule.22) Co-amorphouses of course have an enhanced solubility and dissolution rate because of their higher energy state resulting from amorphization. Since a synthon is a molar stoichiometric unit, the amount needed of a counter molecule used as a drug additive is generally less than that of a polymer in a solid dispersion. Co-amorphouses can bring smaller size/volume and the relatively lower cost by the individual components. Compared with co-crystals, co-amorphouses do not require a crystallization process and give solubility enhancement by the amorphous state. Co-amorphouses show certain advantages as novel formulation technologies. However, the feasibility of co-amorphouses as drug substance or components in drug products has not been fully clarified. And, from the aspects of the functional mechanism and properties, the relationships between the interactions and the physicochemical properties also have not been investigated in detail.
We investigated the physicochemical properties of the co-amorphouses of itraconazole (ITZ)-fumaric acid (FA) and ITZ-L-tartaric acid (TA) pairs prepared by mechanical grinding. The chemical structures are shown in Fig. 1. Itraconazole is an anti-fungal drug having extremely low aqueous solubility. The both drug molecule-counter molecule pairs can form the co-crystals with a molar ratio of ITZ : counter=2 : 1 that improve the solubility of drug in a previous study.23,24) In this study, we discussed the synthon interactions in the co-amorphouses and the characteristic properties depending on the difference of counter molecule, especially their stability, and evaluated their relevancy on drug development in comparison with the properties of the co-crystals that are composed of the same drug-counter molecule pairs.
ITZ was purchased from Spectrum Chemical Manufacturing Corp. (New Brunswick, NJ, U.S.A.). FA, TA, and all other reagents were purchased from Wako Pure Chemical Industries, Ltd. (Osaka, Japan). All purchased materials were reagent grade.
Prepration of the MaterialsPreparation of the Physical MixturesITZ and each counter molecule were gently mixed at a molar ratio of 2 : 1 using an agate mortar and pestle on a 300-mg weighing scale.
Preparation of the Co-crystalsITZ and each counter molecule (molar ratio=2 : 1) were dissolved in tetrahydrofuran (THF) at about 40 mg/mL and at 60°C. The solution was filtered through a 0.45 mm cartridge filter and an equimolar amount of 1-heptane (60°C) was added. The solution was gently cooled to 5°C under stirring at 300 rpm. The precipitates were collected on a filter membrane by vacuum aspiration.
Preparation of the Co-amorphouses by GrindingITZ and each counter molecule (molar ratio=2 : 1) were ground on a 300-mg weighing scale in a 35-mL stainless steel cell containing a 15-mm radius stainless steel by Retsch MM301 vibration ball mill (Haan, Germany) for 15 min. Before grinding, 0.3 mL of 1-heptane was added into the cell. The ground powders were dried by vacuum at room temperature overnight.
Preparation of the Amorphous Materials by LyophilizationITZ and each counter molecule (molar ratio=2 : 1) were dissolved in dimethylformamide at about 100 mg/mL. Twice volume of tert-butyl alcohol was added to the solution and the solution was subsequently frozen at −40°C. The frozen material was lyophilized in an EYELA freeze drier (Tokyo, Japan).
Analytical Procedures for CharacterizationPowder X-Ray DiffractionPowder X-ray diffraction (PXRD) was performed using a Rigaku Rint Ultima IV diffractometer (Akishima, Japan). The X-ray tube voltage and current was 40 kV and 50 mA, respectively. The measurement angle ranged from 2°2θ to 35°2θ. The detection interval was 0.02°2θ.
Raman SpectrometryRaman spectroscopy was performed using a Kaiser Raman Rxn1™ Analyzer (Ann Arbor, MI, U.S.A.). The laser wavelength and power was 785 nm and 400 mW, respectively. About 1 mg of sample was measured on the device’s glass slide. The spectra were acquired at a 4 cm−1 spectral width and 4 s exposures.
Thermal AnalysisDifferential scanning calorimetry (DSC) was performed using a Mettler-Toledo DSC1 (Greifensee, Switzerland). The sample amount used was 0.5 mg. The DSC measurements were performed in a closed aluminum crucible. The temperature range was 25°C–200°C with a ramp rate of 10°C/min.
Dynamic Vapor SorptionDynamic vapor sorption analysis was conducted by a TA VTI SGA-100 (Milford, MA, U.S.A.). The sample amount was about 5 mg. The relative humidity (RH) increased from 5 to 95% with a 5% interval during adsorption and decreased from 95 to 5% with a 5% interval during desorption. The sample temperature was maintained at 25°C.
HPLC AnalysisHPLC analysis was conducted using a Waters Alliance 2695 HPLC system equipped with a 2996 Photo-diode array detector (Milford, MA, U.S.A.). The analytical column was a Shiseido CapcellPak MGIII (3 µm particle size, 4.6 mm inner diameter, and 75 mm length) (Kyoto, Japan). The mobile phases (MP) consisted of 50 mM ammonium acetate solution (MP-A) and acetonitrile (MP-B). The flow rate, column temperature, detection wavelength, and injection volume was 1.0 mL/min, 40°C, 254 nm, and 10 µL, respectively. The mobile phase gradient was MP-B 20% (0.0 min) to 20% (4.0 min) to 80% (10.0 min) to 80% (14.0 min) to 20% (14.1 min) to 20% (20.0 min). The total run time was 20 min, the ITZ sample concentration was 0.2 mg/mL as ITZ free base, and the dissolving solvent was acetonitrile.
Testing Procedures of Physicochemical EvaluationStability Testing ConditionsThe stability testing conditions were performed in the following cycle: at 2–8°C with the container closed, at 25°C/58% RH with the container opened, and at 40°C/75% RH with the container opened. The 2–8°C condition cycle was performed in a refrigerator and the 25°C/58% RH and 40°C/75% RH conditions were performed in a temperature-controlled chamber. The RH was controlled using salt-saturated water, 58% RH by sodium bromide and 75% RH by sodium chloride. The initial and stored materials were analyzed by PXRD and HPLC, respectively. Hygroscopicity of the materials was evaluated by dynamic sorption analysis.
Solubility TestingAbout 2 mg of sample was placed in a glass tube. After addition of 1 mL of the medium, the tube was mixed on a vortex mixer for 30 s. The suspension was shaken at 100 rpm at 37°C for 30 min. The suspension was filtered through a Merck Millipore Millex-GV 13-mm cartridge filter (Darmstadt, Germany). The testing number was n=3. The concentration of the filtrates was quantified using 0.1 mg/mL extra concentration standard by HPLC, Shimadzu UFLC Prominence (Kyoto, Japan). The HPLC conditions were as described previously.25)
Figure 2(A) shows the PXRD patterns of the ground materials of ITZ-FA and ITZ-TA. The patterns of the ground materials of both pairs were represented by halo patterns without diffracted sharp peaks, indicating that the materials were amorphous. For both the ITZ-FA and ITZ-TA pair, very small broad peaks were identified at a low angle range (<10°2θ) in the pattern. The peaks are thought to be derived from the remaining of each co-crystal. The magnified portion of low angle region of PXRD pattern is shown in Fig. 2(C). Taking into account the intensity of the small peaks, the remaining amount was minor.

(A) Initial samples: A, ITZ-TA co-amorphous; B, ITZ-FA co-amorphous; C, ITZ-TA co-crystal; D, ITZ-FA co-crystal; E, TA; F, FA; G, ITZ. (B) Stored samples: A, ITZ-TA lyophillized 40°C 75%RH 4W; B, ITZ-FA lyophillized 40°C 75%RH 4W; C, ITZ-TA ground co-amorphous 40°C 75%RH 4W; D, ITZ-FA ground co-amorphous 40°C 75%RH 4W; E, ITZ-TA co-crystal; F, ITZ-FA co-crystal; G, ITZ. (C) A magnified portion of PXRD patterns at low angle region (<=10°2θ), Co-C: co-crystal, Co-A: co-amorphous, Lyo: lyophilized material, ini: initial, 4W: 40°C/75%RH 4W storage.
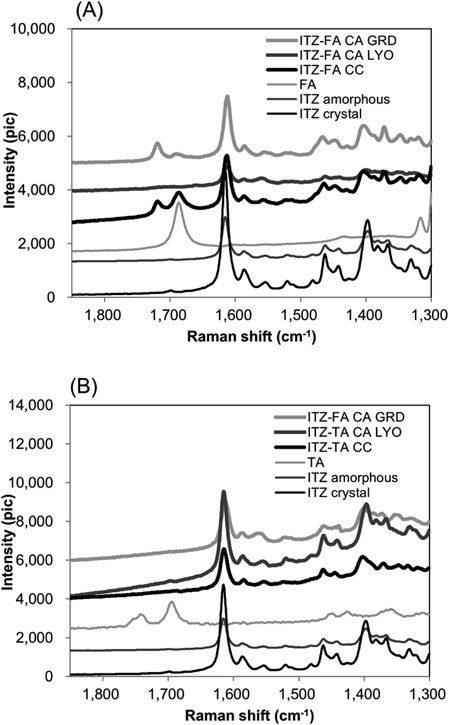
The Raman spectra of all materials are shown in Fig. 3. The lyophilized materials of ITZ-FA and ITZ-TA were prepared and measured to compare the properties as reference amorphous mixtures that were composed of the same drug and counters. For the ITZ-FA materials, a strong peak appeared at 1720 cm−1 in the spectra of the ground co-amorphous. In the lyophilized material, the peak was not identified strongly. The peak was also observed in the spectra of ITZ-FA co-crystal. In the co-crystal spectrum, the peak was observed as a result of the interaction between the drug and the counter (the synthon), which were not seen for the single drug and the counter. As discussed in previous study,26) the peak was derived from the carbonyl group of COOH in FA, which was shifted to a higher wavenumber by an interaction between OH of COOH in FA and 1, 2, 4-triazole group in ITZ, from 1687 cm−1 (on single FA) to 1720 cm−1 (on co-crystal and co-amorphous). The peak at 1720 cm−1 in the co-amorphous spectrum partly included the peak from the co-crystal remaining. However, the amount of remaining of co-crystal was relatively very minor from the PXRD pattern. The peak was believed to be mainly derived from the interaction in the co-amorphous, same as the co-srystal. Therefore, the peak in the co-amorphous spectrum reflected the interaction between ITZ and FA in the amorphous state. In contrast, no peaks were observed in the region in the spectra of the ground and lyophilized amorphous ITZ-TA, even in the co-crystal. The spectra of the ground co-amorphous and lyophilized materials were almost identical to those of the single amorphous ITZ formulation. For ITZ-TA pair, it is supposed that the interaction between ITZ and TA is weak or not-constant.
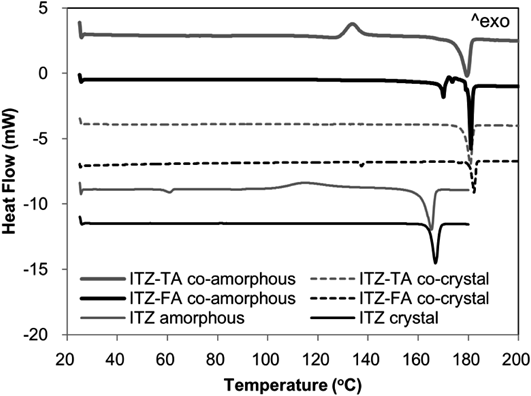
The DSC curves of all amorphous materials are shown in Fig. 4. Amorphous ITZ showed a thermal behavior indicative of glass transition with endothermic peak by enthalpy relaxation at ca. 58°C, an exothermic crystallization peak at ca. 115°C, and a large endothermic peak reflecting the melting of the ITZ crystal at ca. 165°C. ITZ-FA co-amorphous showed no clear thermal behaviors from room temperature to around 170°C but crystallization to the co-crystal and melting as the co-crystal form at 170 and 180°C, respectively. Similarly, ITZ-TA co-amorphous had no clear thermal behaviors from room temperature to around 135°C but crystallization and melting as the co-crystal at 135 and 175°C, respectively. The glass transition was not clearly identified in both co-amorphouses due to a lack of sensitivity of the instrument or the possibility of the ground co-amorphouses not having a clear glass transition behavior like conventional amorphouses by structural disorder during grinding. The crystallization of both co-amorphouses to the co-crystals at over 130°C was caused by the interaction between ITZ and FA or TA that prevents the crystallization to ITZ single crystal.
Overall, the results showed that the interactions between ITZ and the counters were present in the co-amorphous materials, strongly in ITZ-FA pair.
Physical Stability of the Co-amorphous CompoundsTo evaluate the physical stability of the co-amorphouses, the crystallinity and crystal form after storage were evaluated by PXRD. The PXRD patterns of ITZ-FA and ITZ-TA co-amorphouses after one to four weeks of storage are shown in Fig. 2(B). Table 1 shows the solid states of the samples after storage as determined by PXRD. Both ground ITZ-FA co-amorphous and ITZ-TA co-amorphous had remained in the amorphous state even after four weeks of storage at 40°C/75% RH. The PXRD pattern of ITZ-FA and ITZ-TA co-amorphouses had not mostly changed with respect to the storage conditions and time.
| Counter | Preparation | Storage conditon | Period | ||||
|---|---|---|---|---|---|---|---|
| Initial | 1W | 2W | 4W | 8W | |||
| ITZ-FA | Lyophyllized | 2–8°C close | Amorphous | Co-crystal | Co-crystal | Co-crystal | Co-crystal |
| Ground | Amorphous | Amorphous | Amorphous | Amorphous | Amorphous | ||
| Lyophyllized | 25°C 58%RH open | Amorphous | Co-crystal | Co-crystal | Co-crystal | Co-crystal | |
| Ground | Amorphous | Amorphous | Amorphous | Amorphous | Amorphous | ||
| Lyophyllized | 40°C 75%RH open | Amorphous | Co-crystal | Co-crystal | Co-crystal | Co-crystal | |
| Ground | Amorphous | Amorphous | Amorphous | Amorphous | Amorphous | ||
| ITZ-TA | Lyophyllized | 2–8°C close | Amorphous | ITZ | ITZ | ITZ | ITZ |
| Ground | Amorphous | Amorphous | Amorphous | Amorphous | Amorphous | ||
| Lyophyllized | 25°C 58%RH open | Amorphous | ITZ | ITZ | ITZ | ITZ | |
| Ground | Amorphous | Amorphous | Amorphous | Amorphous | Amorphous | ||
| Lyophyllized | 40°C 75%RH open | Amorphous | ITZ | ITZ | ITZ | ITZ | |
| Ground | Amorphous | Amorphous | Amorphous | Amorphous | Amorphous | ||
For both co-amorphouses, slight increase of small peaks in low angle region (<10°2θ) was observed at 40°C/75%RH. Since the small peaks were thought to be the remaining of the co-crystal during the grinding procedure, the slight crystal growth of the co-crystals arose on the high temperature and moisture storage. The magnified portion of PXRD patterns were shown in Fig. 2(C).
The lyophilized amorphous compounds as the reference of co-amorphous were crystallized under all storage conditions tested, suggesting that they were not as physically stable as the co-amorphouses. The crystal form of the crystallized amorphous compounds after storage was different. Lyophilized amorphous ITZ-FA was crystallized to ITZ-FA co-crystal, while lyophilized amorphous ITZ-TA was crystallized to a single ITZ free base crystal. The crystallization behavior might be associated with the strength of the interaction between ITZ and the counters. The ITZ-FA interaction in the lyophilized material was enough strong and of the same structure as that of the synthon of ITZ-FA co-crystal that is formed between ITZ and FA, resulting in the crystallization to the co-crystal. In lyophilized amorphous ITZ-TA, the interaction between ITZ and TA was weaker than that between the ITZ molecules since no synthon was formed in the co-crystal, resulting in the crystallization as ITZ free base.
The volume and surface area of the lyophilized material were usually larger than those of the ground co-amorphous. The mobility and accessibility of the lyophilized amorphous material to water were, therefore, higher and the lyophilized amorphous material was more readily crystallized, especially under heat and moist conditions. The mechanical grinding force used to prepare the ground co-amorphous might have stabilized the amorphous state by promoting the interaction between ITZ and the counter molecules (FA, TA), thereby providing a lower enthalpy. Furthermore, mechanical force causes long-range structural disorder in ground materials, which increases the entropy. A ground material is believed to cause a local minimum in the Gibbs energy and, hence, has a lower Gibbs energy than a lyophilized amorphous material. As shown in the Raman spectra of the lyophilized amorphous compounds, the spectra are the same as those of ITZ free base. Oppositely, the peak at 1720 cm−1 observed in the Raman spectra of the ITZ-FA ground co-amorphous indicated the presence of an interaction between ITZ and FA.
Lyophilization is an effective technology to produce amorphous drug materials as seen from the perspective of drug product manufacturing. However, our results of the physical stability pointed out that it might not be suitable for producing co-amorphous as issues relevant to the pharmaceutical industry. These days, the produce of co-amorphous by spray-drier was reported,27) indicating the scalability of co-amorphouses became available.
Chemical Stability of the Co-amorphous CompoundsThe chemical stability of the co-amorphouses was evaluated by the appearance (mainly color) and the chemical purity based on the amount of related substances measured by reversed-phase HPLC. Table 2 lists the main peak area percentage of the ITZ-FA and ITZ-TA chromatograms after storage, respectively. The lyophillized materials, co-crystals and physical mixtures used as references were stored under the same conditions, the results of which are included in the Table.
| Counter | Preparation | Storage conditon | Purity by main peak area (%) | |||
|---|---|---|---|---|---|---|
| Period | ||||||
| Initial | 1W | 2W | 4W | |||
| ITZ-FA | Lyophyllized | 2–8°C close | 99.6 | 99.6 | 99.6 | 99.6 |
| Ground | 99.6 | 99.6 | 99.5 | 99.5 | ||
| Physical mixture | 99.6 | 99.6 | 99.6 | 99.6 | ||
| Co-crystal | 99.6 | 99.6 | 99.6 | 99.6 | ||
| Lyophyllized | 25°C 58%RH open | 99.6 | 99.6 | 99.6 | 99.6 | |
| Ground | 99.6 | 99.6 | 99.5 | 99.5 | ||
| Physical mixture | 99.6 | 99.6 | 99.6 | 99.6 | ||
| Co-crystal | 99.6 | 99.6 | 99.6 | 99.6 | ||
| Lyophyllized | 40°C 75%RH open | 99.6 | 99.6 | 99.6 | 99.6 | |
| Ground | 99.6 | 99.6 | 99.5 | 99.5 | ||
| Physical mixture | 99.6 | 99.6 | 99.6 | 99.6 | ||
| Co-crystal | 99.6 | 99.6 | 99.6 | 99.6 | ||
| ITZ-TA | Lyophyllized | 2–8°C close | 99.6 | 99.6 | 99.6 | 99.6 |
| Ground | 99.6 | 99.6 | 99.5 | 99.5 | ||
| Physical mixture | 99.6 | 99.6 | 99.6 | 99.6 | ||
| Co-crystal | 99.6 | 99.6 | 99.6 | 99.6 | ||
| Lyophyllized | 25°C 58%RH open | 99.6 | 99.4 | 99.1 | 98.8 | |
| Ground | 99.6 | 99.6 | 99.5 | 99.5 | ||
| Physical mixture | 99.6 | 99.6 | 99.6 | 99.6 | ||
| Co-crystal | 99.6 | 99.6 | 99.6 | 99.6 | ||
| Lyophyllized | 40°C 75%RH open | 99.6 | 99.2 | 98.6 | 98.1 | |
| Ground | 99.6 | 99.6 | 98.7 | 95.6 | ||
| Physical mixture | 99.6 | 98.9 | 98.9 | 97.7 | ||
| Co-crystal | 99.6 | 99.6 | 99.6 | 99.6 | ||
The color of all compound materials present in the ITZ-FA pair was not clearly changed. In contrast, the color of the ITZ-TA pair was changed to a reddish and brownish shade, except for that of ITZ-TA co-crystals (data not shown). The color change was indicative of degradation.
The amount of the related substances present in both ITZ-FA pairs did not increase under all storage conditions tested. FA did not react with ITZ as was observed in the physical mixture. The ground ITZ-FA co-amorphous was also chemically stable. The amorphization and the interaction formed by the synthon did not affect the reactivity of FA with ITZ. ITZ-FA was physically and chemically stable, suggesting the potential of this novel solid drug form or of the intermediate drug products for the industrial development of ITZ.
On the other hand, the related substances present in ground ITZ-TA co-amorphous increased with 4.0% after storage at 40°C/75% RH for four weeks, similar to what was observed for the lyophilized amorphous and physical mixture. Since the related substances had a lower lipophilicity than that of ITZ as inferred from their faster retention time compared with that of ITZ on reverse-phase HPLC testing, it is speculated that the degradation products have been generated by the oxidation or hydroxylation of the ITZ molecules. Although TA was chemically reactive with ITZ, only the ITZ-TA co-crystal was stable in the ITZ-TA pairs. Crystallization as the co-crystal fixed the TA molecule in the crystal lattice and decreased the mobility of TA to react with ITZ. ITZ and TA interacted within the co-amorphous compound, even though the interaction in ITZ-TA co-amorphous was weaker than that in ITZ-FA co-amorphous. It was expected that this interaction would decrease the reactivity of TA and chemically stabilize the co-amorphous. However, the interaction was not enough strong to decrease the reactivity of TA and, hence, did not contribute to chemical stabilization. Thereby, a co-amorphous compound has intrinsic mobility, which underlies its high reactivity as an amorphous material.
When selecting the counter molecule for co-amorphous to improve the physicochemical properties of a drug, the counter molecules should not be intrinsically reactive with the drug molecules. In the ITZ-TA system, the interaction within the co-amorphous may not be enough strong to prevent the reaction of the counter molecules with the drug molecules. Therefore, in such cases, a co-crystal screening strategy is more applicable than a co-amorphous approach.
The hygroscopicity of both co-amorphous was determined using a dynamic vapor sorption analyzer, providing interesting results regarding the properties of the amorphous material. The RH and weight change plots of ITZ-FA and ITZ-TA are shown in Figs. 5(A) and (B), respectively. Most amorphous materials are hygroscopic because of the large surface area, high surface free energy, and the high mobility, all of which enhance water absorption. However, ITZ-FA co-amorphous was not hygroscopic, in contrast to ITZ-TA, which was highly hygroscopic as expected from an amorphous material, showing a 9% weight change as a result of the change in RH. The interaction between ITZ and FA is strong and is mainly composed of hydrogen bonds. This interaction folds the hydrophilic moiety, thereby sterically hindering the exposure of hydrophilic molecules into the amorphous material. The surface of ITZ-FA is hydrophobic, preventing water molecules to access the hydrophilic parts in the co-amorphous compound. Due to this strong interaction, the mobility of the entire ITZ-FA co-amorphous material is low and the relative surface area is not enough large for water molecules to attach to the surface. The interaction in ITZ-TA co-amorphous, on the other hand, is weak, the mobility is high, and the hydrophilic parts of the ITZ molecules, especially the hydroxyl groups, and of the TA molecules are most likely exposed to the surface of the co-amorphous compound. Water molecules have free access to the hydrophilic parts and can adsorb to the surface of ITZ-TA co-amorphous. This water uptake property of ITZ-TA co-amorphous also contributed to the higher degradation observed under a humid condition of 40°C/75% RH than that observed for the physical mixture.

Table 3 shows the solubility for 1% hydroxy propyl methyl cellulose (HPMC) in the first fluid of distinregration test in Japanese pharmacopoeia XVI (JP1), simulating the fluid in stomach. ITZ-FA and ITZ-TA co-amorphouses were more soluble than ITZ free base crystal. The solubility of both co-amorphouses was almost the same as that of the ITZ amorphous compound. The co-amorphouses also had higher solubilities than those of the co-crystals, whose solubility was much higher than that of ITZ free base. Since the co-amorphous compound did not contain a crystal lattice, it was more soluble than the co-crystal.
| Solubility (µg/mL, n=3, Average±S.D.) | |
|---|---|
| ITZ crystal | 4.2±0.1 |
| ITZ amorphous | 165.3±15.6 |
| ITZ-FA co-crystal | 28.8±2.0 |
| ITZ-TA co-crystal | 129.2±5.3 |
| ITZ-FA co-amorphous | 149.4±1.4 |
| ITZ-TA co-amorphus | 203.2±4.2 |
It is thought that the ITZ-FA interaction is tight and rigid; conversely, the ITZ-TA interaction is loose and flexible. A reactant can interfere with the interaction of ITZ-TA, which has a relatively higher energy state coupled to a high degree of reactivity and can, therefore, react with ITZ molecules. To elucidate the different strengths of the interaction, the acid dissociation constant, pKa, is often used. However, the calculated pKa of FA and TA is almost identical and cannot explain their interaction strength difference. The interaction strength is not the result of the electrostatic effect. Probably, FA’s and TA’s steric effect may largely affect the interaction strength. TA has a larger structure volume and structure flexibility than FA whose main backbone consists of a carbon–carbon double bond providing FA with a more flat structure, suggesting the interactions between ITZ and FA are tighter and more rigid than those between ITZ and TA. And, TA has two OH groups connecting to 2- and 3-position in the structure that can serve as a proton donor. It is likely that the intramolecular interaction between 1-COOH and 3-OH/2-COOH and 4-OH form in the ITZ-TA co-crystal and co-amorphous. The intramolecular interaction in TA may weaken the intermolecular interaction between COOH in TA and 1, 2, 4-triazole ring in ITZ, presuming another reason of weaker interaction in ITZ-TA pair. Therefore, a clear Raman shift caused by the interaction was not observed in the Raman spectra of ITZ-TA co-crystal and co-amorphous.
Comparison between the Co-crystal and Co-amorphous CompoundsFor both ITZ-FA and ITZ-TA, the co-amorphous state was more soluble than the free base crystal. ITZ-FA co-amorphous was also more soluble than the co-crystal and physically and chemically stable like as the amorphous solid dispersion formulation with hydrophilic polymers. ITZ-FA co-amorphous was superior to the co-crystal regarding its solubility and could be developed into a drug. Another advantage is that in the production of a co-amorphous compound the crystallization of the co-crystal is not required. Therefore, if a co-amorphous compound can be prepared by standard manufacturing processes, the implementation of the co-amorphouses in drug products may be relevant for drug development. ITZ-TA co-amorphous was more soluble but less stable than the co-crystal. The co-crystal could, therefore, be better implemented in the ITZ-TA pair instead of the co-amorphous. However, the development of the co-crystal does not proceed without disadvantages: if the co-crystal loses its crystallinity during the storage and formulation process, its chemical stability decreases to a degree similar to that of the physical and the co-amorphous.
We investigated the physicochemical properties of ground co-amorphous of itraconazole with organic acids and compared the properties with the co-crystals having the same drug–counter pairs. We successfully identified the developability of ITZ-FA co-amorphous on chemical/physical stability and suggested the merit for the co-crystal on the higher solubility. It is confirmed that a co-amorphous approach is one of the effective technologies to improve physicochemical properties of drugs in concurrence with a co-crystal approach.
The authors gratefully acknowledge all members in Pharmaceutics Research, Analytical Development Laboratories, CMC Center, Takeda Pharmaceutical Co., Ltd., for their insight and dedicated support. We also would like to thank Mr. Steven Glenn, Head of Analytical Development Laboratories, CMC Center, Takeda Pharmaceutical Co., Ltd., for kind assistance and discussion
Authors are employees of Takeda Pharmaceutical Company Ltd.