Experimental
Melting points were determined using a micro melting point apparatus (Yanaco MP-S3) without correction. IR spectra were measured by a Shimadzu FTIR-8100 IR spectrophotometer. Low- and high-resolution mass spectra (LR-MS and HR-MS) were obtained by a JEOL JMS HX-110 double-focusing model equipped with an FAB ion source interfaced with a JEOL JMA-DA 7000 data system. lH- and 13C-NMR spectra were obtained by JEOL JNM A-500 and ECG600R (only for compounds 7cm, 9d and 16o). Chemical shifts were expressed in δ ppm downfield from an internal TMS signal for lH-NMR and the carbon signal of the corresponding solvent [CDCl3 (77.00 ppm), dimethyl sulfoxide (DMSO)-d6 (39.50 ppm)] for 13C-NMR. The abbreviations dd=double doublets, dt=double triplets, and dm=double multiplets are used for the multiplicity of lH-NMR data. The signal assignments were confirmed by two-dimensional (2D)-NMR analyses: lH–lH 2D correlation spectroscopy (COSY), lH–l3C heteronuclear multiple-quantum coherence (HMQC), and lH–l3C heteronuclear multiple-bond connectivity (HMBC). Microanalyses were performed with a Yanaco MT-6 CHN corder. Routine monitoring of reactions was carried out using precoated Kieselgel 60F254 plates (E. Merck). Open and flash column chromatography or centrifugal chromatography separations of the reaction products were performed on silica gel (Kanto 60N or Able-Biott) with a UV detector. Commercially available starting materials were used without further purification, and dry solvents were used in all reactions.
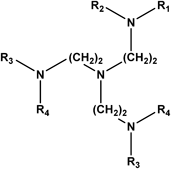
General Procedure for the Preparation of C3-Symmetrical Amine-Type TAEA Derivatives (3): Example: Synthesis of 4,4′,4″-[Nitrilotris(2,1-ethanediyliminomethylene)]tris(2-methoxyphenol) (3b) (Entry 2) (Step 1)To a solution of vanillin (2b, 2.28 g, 15.0 mmol) in methanol (MeOH, 6 mL) was added TAEA (1, 731 mg, 5.00 mmol) in MeOH (3 mL) at room temperature (r.t.) under an N2 atmosphere. After stirring for 1 h, the yellow solution changed to a yellow sticky suspension.
(Step 2)After dilution with MeOH (12 mL) and ice-cooling, compound 1 (1.84 g, 10.0 mmol) in dry benzene (20 mL) was added dropwise with stirring. After stirring for 20 min at r.t., the reaction mixture was refluxed for 1 h. After cooling to r.t., a small amount of NaBH4 (945 mg, 25.0 mmol) was added to the mixture and the mixture was stirred for 2.5 h at 0°C. It was then diluted with aqueous ammonium chloride (NH4Cl, 10%, 100 mL) and extracted with CH2Cl2 (3×200 mL). The combined organic layer was washed with brine (50 mL) and dried over anhydrous magnesium sulfate (MgSO4). Evaporation of the solvent gave crude 3b (2.43 g, 88%). An analytical sample of 3b was obtained by recrystallization from chloroform (CHCl3) as an off-white solid.
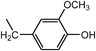
3b: mp 64–69°C (from CHCl3). IR (KBr) cm−1: 1279, 1034, 1125 (C–O). 1H-NMR (DMSO-d6) δ: 2.5–2.7 (12H, m, H1′, 2′), 3.21 (6H, br s, NH, OH), 3.54 (6H, br s, Hα), 3.71 (9H, br s, OCH3), 6.60–6.73 (6H, m, H5, 6), 6.84 (3H, d, J=1.5 Hz, H3), 8.30 (<1H, s, CHCl3). 13C-NMR (DMSO-d6) δ: 46.56 (C1′), 52.75 (Cα), 54.00 (C2′), 55.44 (OCH3), 79.08 (CHCl3), 112.07 (C3), 114.99 (C6), 120.14 (C5), 131.60 (C4), 145.09 (C1), 147.31 (C2). Positive-ion FAB-MS m/z: 555 (M+H+). HR-FAB-MS m/z: 555.3185 (Calcd for C30H43N4O6: 555.3183). Anal. Calcd for C30H42N4O6·0.875CHCl3: C, 56.26; H, 6.56; N, 8.50. Found: C, 56.29; H, 6.62; N, 8.45.
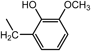
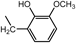
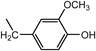
2,2′,2″-[Nitrilotris(2,1-ethanediyliminomethylene)]tris[6-methoxyphenol] (3a) (Entry 1)Compound 3a was prepared from the reaction of 1 with o-vanillin (2a) under the conditions shown in Table S1. Separation of the products by centrifugal chromatography (CH2Cl2 : 95% EtOH : 28% NH3=90 : 9.5 : 0.5→85 : 13 : 2) gave 3a (1.73 g, 62%) as a reddish purple solid.
3a: mp 35–38°C (mp 42–45°C).15) IR (KBr) cm−1: 3395 (OH), 3305 (NH), 1590, 1475 (C=C of Ar), 1235, 1075 (C–N and C–O). 1H-NMR (CDCl3) δ: 2.55 (6H, m, H2′), 2.68 (6H, m, H1′), 3.83 (9H, br s, CH3O–), 3.97 (6H, br s, Hα), 6.27 (6H, br s, NH, OH), 6.60 (3H, d, J=7.6 Hz, H3), 6.71 (3H, t, J=7.6 Hz, H4), 6.78 (3H, d, J=7.6 Hz, H5). 13C-NMR (CDCl3) δ: 45.78 (C1′), 51.76 (Cα), 53.90 (C2′), 55.76 (CH3O–), 110.75 (C5), 118.52 (C4), 120.69 (C3), 122.69 (C2), 147.10 (C1), 147.83 (C6). Positive-ion FAB-MS m/z: 555 (M+H)+. HR-FAB-MS m/z: 555.3202 (Calcd for C30H43N4O6: 555.3183). Anal. Calcd for C30H42N4O6·H2O: C, 62.92; H, 7.74; N, 9.78. Found: C, 62.93; H, 7.71; N, 9.57.
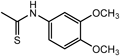
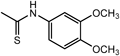
N2-[(3,4-Dimethoxyphenyl)methyl]-N1,N1-bis[2-[[(3,4-dimethoxyphenyl)methyl]amino]ethyl]-1,2-ethanediamine (3c) (Entry 3)Compound 3c was prepared from the reaction of 1 with 3,4-dimethoxybenzaldehyde (2c) under the conditions shown in Table S1. Separation of the products by flash chromatography (CH2Cl2 : 95% EtOH : 28% NH3=93 : 6.6 : 0.4→85 : 14 : 1) gave 3c (5.00 g, 84%) as a sticky yellow oil.
3c: 1H-NMR (CDCl3) δ: 2.75–2.77 (12H, m, H1′, H2′), 3.84 (6H, s, Hα), 3.85 (9H, s, OCH3 on C3), 3.90 (9H, s, OCH3 on C4), 4.36 (3H, br s, NH), 6.79 (3H, d, J=8.2 Hz, H5′), 6.86 (3H, dd, J=8.2, 2.1 Hz, H6′), 7.13 (3H, d, J=2.1 Hz, H2′). 13C-NMR (CDCl3) δ: 45.21 (C1′), 52.24 (Cα), 53.02 (C2′), 55.92 (OCH3 on C4), 56.22 (OCH3 on C3), 111.16 (C5), 112.58 (C2), 121.45 (C6), 127.88 (C1), 149.02 (C3), 149.39 (C4). Positive-ion FAB-MS m/z: 597 (M+H+). HR-FAB-MS m/z: 597.3660 (Calcd for C33H49N4O6: 597.3652). Anal. Calcd for C33H48N4O6·2.4H2O: C, 61.93; H, 8.32; N, 8.45. Found: C, 61.99; H, 8.17; N, 8.72.
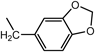
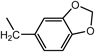
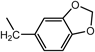
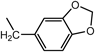
N2-(1,3-Benzodioxol-5-ylmethyl)-N1,N1-bis[2-[(1,3-benzodioxol-5-ylmethyl)amino]ethyl]-1,2-ethanediamine (3d) (Entry 4)Compound 3d was prepared from the reaction of 1 (1.46 g, 10.0 mmol) with piperonal (2d) under the conditions shown in Table S1. Separation of the products by centrifugal chromatography (CH2Cl2 : 95% EtOH : 28% NH3=85 : 14 : 1) gave 3d (0.760 g, 14%) as a pale yellow oil.
(Entry 5)This compound was also prepared as follows: To a solution of 2d (2.25 g, 15.0 mmol) in benzene (40 mL) were added compound 1 (731 mg, 5.0 mmol) and p-toluenesufonic acid (TsOH, 66 mg, 0.35 mmol) and the resulting mixture was refluxed. The resultant H2O was removed with a Dean–Stark distillation apparatus for 18 h under an N2 atmosphere. After evaporation of the solvent, the resulting mixture was diluted with MeOH (30 mL) and then a small amount of NaBH4 (1.57 mg, 41.5 mmol) was added and the mixture was stirred for 3 h at 0°C under an N2 atmosphere. After evaporation of the solvent, CHCl3 (20 mL), brine (5 mL), and 5% NaHCO3 (5 mL) were added to the obtained yellow solid, and then the resulting mixture was vigorously stirred for 1 h. After separation of the organic layer, the aqueous layer was re-extracted with CHCl3 (2×20 mL). The combined organic layer was dried over MgSO4, and evaporation of the solvent gave a brown oil. The product was purified by flash chromatography (CH2Cl2 : 95% EtOH : 28% NH3=73 : 25 : 2→65 : 32 : 3) to give 3d (1.38 g, 50%) as a yellow oil. An analytical sample of 3d was obtained as a pale yellow solid by centrifugal chromatography (CH2Cl2 : 95% EtOH : 28% NH3=80 : 19.5 : 0.5).
3d: mp 215°C (dec.). IR (NaCl) cm−1: 3300 (NH of amine), 2825 (CH2 of methylenedioxy), 1245, 1040 (=C–O–C–), 930 (C–O). 1H-NMR (DMSO-d6) δ: 2.76 (6H, t, J=5.2 Hz, H2′), 3.06 (6H, t, J=5.2 Hz, H1′), 4.11 (6H, s, Hα), 6.04 (6H, s, H2), 6.93 (3H, d, J=7.9 Hz, H7), 7.07 (3H, dd, J=7.9, 1.5 Hz, H6), 7.27 (3H, d, J=1.5 Hz, H4), 9.48 (3H, br s, NH). 13C-NMR (DMSO-d6) δ: 43.28 (C1′), 49.54 (C2′ or Cα), 49.62 (Cα or C2′), 101.16 (C2), 108.13 (C7), 110.42 (C4), 124.24 (C6), 125.14 (C5), 147.22 (C3a), 147.58 (C7a). 1H-NMR (CDCl3) δ: 2.17 (3H, br s, NH), 2.56 (6H, t, J=5.8 Hz, H2′), 2.64 (6H, t, J=5.8 Hz, H1′), 3.64 (6H, s, Hα), 5.90 (6H, s, H2), 6.70 (6H, br s, H6, 7), 6.79 (3H, s, H4). 13C-NMR (CDCl3) δ: 46.88 (C1′), 53.57 (Cα), 54.29 (C2′), 100.76 (C2), 107.95 (C7), 108.55 (C4), 121.08 (C6), 134.14 (C5), 146.41 (C7a), 147.62 (C3a). Positive-ion FAB-MS m/z: 549 (M+H)+. HR-FAB-MS m/z: 549.2728 (Calcd for C30H37N4O6: 549.2713). Anal. Calcd for C30H36N4O6·0.5NH3: C, 64.67; H, 6.78; N, 11.31. Found: C, 64.42; H, 6.81; N, 11.04.
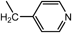
N1-(4-Pyridinylmethyl)-N2,N2-bis[2-[(4-pyridinylmethyl)amino]ethyl]-1,2-ethanediamine (3e) (Entry 6)Compound 3e was prepared from the reaction of 1 with isonicotinaldehyde (2e) under the conditions shown in Table S1. Separation of the products by centrifugal chromatography (CH2Cl2 : 95% EtOH : 28% NH3=73 : 15 : 2→65 : 32 : 3) gave 3e (3.97 g, 95%) as a yellow oil.
3e: 1H-NMR (CDCl3) δ: 2.00 (3H, br s, NH), 2.61 (6H, t, J=5.2 Hz, H2′), 2.67 (6H, t, J=5.2 Hz, H1′), 3.76 (6H, s, Hα), 7.20 (6H, dd, J=4.6, 1.5 Hz, H3, 5), 8.50 (6H, dd, J=4.6, 1.5 Hz, H2, 6). 13C-NMR (CDCl3) δ: 47.17 (C1′), 52.73 (Cα), 54.20 (C2′), 122.78 (C3, 5), 149.33 (C4), 149.80 (C2, 6). KM4-4-1 Positive-ion FAB-MS m/z: 420(M+H+). HR-FAB-MS m/z: 420.2873 (Calcd for C24H34N7: 420.2876).
General Procedure for the Preparation of C3-Symmetrical Amide-Type TAEA Derivatives (5): Example: Synthesis of N,N′,N″-(Nitrilotri-2,1-ethanediyl)tris[N-(3,4-dimethoxyphenylmethyl)acetamide] (5c) (Entry 9)To a solution of compound 3c (2.85 g, 5.00 mmol) in benzene (8 mL) was added dropwise a solution of acetic anhydride (Ac2O, 4, 1.55 g, 15.0 mmol) in benzene (2 mL) at r.t., and then the mixture was stirred for 2 h. After removal of the solvent by evaporation, 1 M NaOH solution (20 mL) was added to the resulting residue and then the mixture was extracted with EtOAc (3×40 mL). The organic layer was washed with brine (25 mL) and dried with MgSO4 and then evaporated to give a pale yellow oil, which was purified by flash chromatography (EtOH : EtOAc=4 : 6) to give 5c (2.88 g, 80%) as a colorless semisolid.
5c: mp 34–39°C. IR (KBr) cm−1: 1646 (C=O), 1261, 1233 (C–O of aromatic ether), 1137, 1025 (C–O of aliphatic ether). 1H-NMR (CDCl3) δ: 2.07, 2.09*, 2.11, 2.15 (9H, s, COCH3), 2.05, 2.52, 2.58*, 2.63 (6H, t, J=7.3 Hz, H2′), 3.17, 3.23, 3.34*, 3.37 (6H, t, J=7.3 Hz, H1′), 3.83, 3.84, 3.86, 3.86* (18H, s, OCH3), 4.39, 4.43*, 4.47, 4.50 (6H, s, Hα), 6.63–6.79 (6H, m, H2, 6), 6.80–6.86 (3H, m, H5). (The signals of the predominant conformer were asterisked.) 13C-NMR (CDCl3) δ: 21.64 (COCH3), 44.28 (C1′), 52.03 (C2′), 52.50 (Cα), 55.98 (OCH3), 109.76 (C2), 111.61 (C5), 118.51 (C6), 129.12 (C1), 148.74 (C3), 149.60 (C4), 170.87 (C=O). (Signals of only the predominant conformer were assigned.) Positive-ion FAB-MS m/z: 723 (M+H+). HR-FAB-MS m/z: 723.3967 (Calcd for C39H55N4O9: 723.3969). Anal. Calcd for C39H54N4O9·H2O: C, 63.22; H, 7.62; N, 7.56. Found: C, 63.18; H, 7.70; N, 7.61.
N,N′,N″-[2,2′,2″-Nitrilotris(ethane-2,1-diyl)]tris[N-(2-hydroxy-3-methoxybenzyl)acetamide] (5a) (Entry 7)Compound 5a was prepared by acylation of 3a with Ac2O (4) under the conditions shown in Table S2. After work-up, crude compound 5a (1.59 g, 75%) was obtained as a white solid. Recrystallization from AcOEt gave an analytically pure product 5a as colorless crystals.
5a: mp 107–109°C (from AcOEt). IR (KBr) cm−1: 3150 (OH), 1625 (C=O), 1070 (C–O of phenol, ether). 1H-NMR (DMSO-d6) δ: 1.98, 1.99, 2.02, 2.03* (9H, s, CH3CO–), 2.36–3.47 (3H, m, H2′), 2.50–2.58 (3H, m, H2′), 3.13–3.24 (6H, m, H1′), 3.74*, 3.78 (9H, s, CH3O–), 4.38, 4.40, 4.42* (6H, br s, Hα), 6.57–6.76 (6H, m, H3, 4), 6.83–6.88 (3H, m, H5), 8.76, 8.79, 8.82, 8.95, 8.96*, 8.98 (3H, br s, OH). (The signals of the predominant conformer were asterisked.) 13C-NMR (DMSO-d6) δ: 20.64, 20.86, 20.90*, 20.93, 21.28, 21.32* (CH3CO–), 43.05, 43.31, 43.37, 43.52, 43.77 (Cα, C1′), 46.16, 46.24*, 46.40 (C1′), 47.39, 47.51* (Cα), 51.07, 51.32*, 51.83, 52.07* (C2′), 55.70*, 55.79* (CH3O–), 110.98, 111.01, 111.10, 111.17*, 111.27 (C5), 118.61, 118.82*, 119.51 (C4), 119.61, 119.79, 121.25* (C3), 123.78, 123.86, 124.09* (C2), 143.99, 144.06, 144.50* (C1), 147.38*, 147.61, 147.64, 147.68 (C6), 169.78, 169.84, 170.43, 170.54*, 170.67 (C=O). (Signals of only the predominant conformer were asterisked.) Positive-ion FAB-MS m/z: 681 (M+H)+. HR-FAB-MS m/z : 681.3517 (Calcd for C36H49N4O9: 681.3500). Anal. Calcd for C36H48N4O9·0.9 H2O: C, 62.04; H, 7.20; N, 8.04. Found: C, 62.09; H, 7.15; N, 7.85.
N,N′,N″-(Nitrilotri-2,1-ethanediyl)tris[N-(4-hydroxy-3-methoxyphenylmethyl)acetamide] Hydrochloride (5b·HCl) and [[(Nitrilotri-2,1-Ethanediyl)tris(acetylazanediyl)]tris(methylene)]tris(2-methoxybenzene-4,1-diyl) Triacetate (5b′) (Entry 8)Compound 5b′ was prepared by acylation of compound 3b (0.943 g, 1.70 mmol) with Ac2O (4, 1.39 g, 13.6 mmol) under the conditions shown in Table S2. After work-up crude compound 5b′ (987 mg, 72%) was obtained as a white solid. After dilution of this material with EtOH (4 mL) and addition of 1 M HCl (in EtOH), evaporation of the solvent and azeotropic evaporation with dry benzene were repeated. Then the white semisolid material was recrystallized from 2-PrOH to give an analytically pure salt of the deacylated target compound 5b·HCl as colorless crystals.
5b·HCl: mp 69–73°C (from 2-PrOH). IR (KBr) cm−1: ca. 3200 (OH), ca. 2600 (NH+), 1636 (C=O), 1279, 1032, 1125 (C–O). 1H-NMR (DMSO-d6) δ: 1.04 (6H, d, J=6.1 Hz, CH3 of 2-PrOH), 2.10*, 2.11 (9H, s, COCH3), 3.17, 3.24*, 3.33 (6H, br s, H2′), 3.39 (4H, br s, OH) 3.66*, 3.75 (6H, br s, H1′), 3.75, 3.76, 3.77*, 3.78 (9H, s, OCH3), 3.80 (1H, q, J=6.1 Hz, CH of 2-PrOH), 4.38, 4.48* (9H, br s, Hα), 6.65* (0.8H, dd, J=7.9, 1.8 Hz, H5), 6.68–6.73 (0.4H, m, H5, 6), 6.79* (0.8H, d, J=7.9 Hz, H6), 6.82* (0.8H, br s, H3), 6.88–6.93 (0.2H, m, H3), 8.96*, 9.85, 10.88, 11.73 (3H, br s, NH, OH). (The signals of the predominant conformer were asterisked.) 13C-NMR (DMSO-d6) δ: 21.40 (COCH3), 25.39 (CH3 of 2-PrOH), ca. 40 (C1′), 50.33 (C2′), 51.29 (Cα), 55.64 (OCH3), 61.93 (CH of 2-PrOH), 111.52 (C3), 115.64 (C6), 119.23 (C4), 127.56 (C5), 146.01 (C1), 147.77 (C2), 171.38 (C=O). (Signals of only the predominant conformer were assigned.) Positive-ion FAB-MS m/z: 681 (M+H+). HR-FAB-MS m/z: 681.3491 (Calcd for C36H49N4O9: 681.3500). Anal. Calcd for C36H48N4O9·HCl·2-PrOH·H2O: C, 58.89; H, 7.48; N, 7.04. Found: C, 58.75; H, 7.70; N, 6.84.
5b′: Positive-ion FAB-MS m/z: 807 (M+H+). HR-FAB-MS m/z: 807.3812 (Calcd for C42H55N4O12: 807.3816). Both 1H- and 13C-NMR in CDCl3 were too complicated and we could not provide full assignments of the observed signals.
N,N',N″-(Nitrilotri-2,1-ethanediyl)tris[N-(1,3-benzodioxol-5-ylmethyl)acetamide] (5d) and N,N′-[[[2-[(1,3-Benzodioxol-5-ylmethyl)amino]ethyl]imino]di-2,1-ethanediyl]bis[N-(1,3-benzodioxol-5-ylmethyl)acetamide] (9d) (Entry 10)Compound 5d was prepared by acylation of 3d with Ac2O (4) under the conditions shown in Table S2. Separation of the products by flash chromatography (CH2Cl2 : 95% EtOH : 28% NH3=95 : 4.7 : 0.3→93 : 6.6 : 0.4) gave 5d (2.67 g, 53%) as a colorless semisolid and 9d (0.94 g, 20%) as a colorless oil. Compound 9d was purified by centrifugal chromatography (EtOAc : EtOH=99.5 : 0.5) to give an analytically pure 9d as a hygroscopic white semisolid.
5d: mp 46–65°C. IR (KBr) cm−1: 1644 (C=O), 1248, 1037 (=C–O–C–), 923 (C–O of methylenedioxy). 1H-NMR (CDCl3) δ: 2.09*, 2.10, 2.11, 2.14 (9H, s, CH3), 2.40–2.70 [6H, 2.43 (br s), 2.49, 2.55* (t, J=7.0 Hz), 2.65 (br s), H2′], 3.10–3.45 [6H, 3.16, 3.22, 3.31* (t, J=7.0 Hz), 3.37 (br s), H1′], 4.36, 4.39*, 4.44, 4.46 (6H, s, Hα), 5.91, 5.91, 5.94*, 5.95, 5.96 (6H, s, H2), 6.57–6.80 (9H, m, ArH). (The signals of the predominant conformer were asterisked.) 13C-NMR (CDCl3) δ: 21.54 (CH3), 44.06 (C1′), 51.84 (C2′), 52.50 (Cα), 101.15 (C2), 106.79 (C4), 108.45 (C7), 119.57 (C6), 130.41 (C5), 147.14 (C3a or 7a), 148.27 (C7a or 3a), 170.71 (C=O). (Signals of only the predominant conformer were assigned.) Positive-ion FAB-MS m/z: 675 (M+H+). HR-FAB-MS m/z: 675.3027 (Calcd for C36H43N4O9: 675.3030). Anal. Calcd for C36H42N4O9·0.7H2O: C, 62.91; H, 6.36 ; N, 8.15. Found: C, 62.90; H, 6.57; N, 8.01.
9d: mp 76–81°C. IR (KBr) cm−1: 1638(C=O), 1251, 1037 (=C–O–C–), 924 (C–O of methylenedioxy). 1H-NMR (DMSO-d6) δ: 1.44 (1H, br s, NH), 1.98*, 1.99 (5H, s, CH3), 2.02 (0.6H, s, CH3), 2.06 (0.4H, s, CH3), 2.29–2.47 (4H, m, H2′), 2.50–2.57 (1H, m, H2″), 2.61–2.69 (1H, m, H2″), 3.05–3.31 (6H, m, H1′, 1‴), 3.45–5.57 (1H, m, Hα′), 3.83–3.89 (1H, m, Hα′), 4.32*, 4.36, 4.39, 4.44 (4H, s, Hα), 5.98*, 5.99 (4H, s, H2), 6.01*, 6.05 (2H, s, H2‴), 6.62–6.70 (2H, m, H6), 6.73–6.77 (2H, m, H4), 6.79–6.92 (4H, m, H7, 6‴, 7‴), 7.03–7.08 (1H, m, H4‴). 13C-NMR (DMSO-d6) δ: 21.50 (CH3), 43.30 (C1″), 45.57 (C1′), 47.55 (Cα), 49.99 (C2″), 51.56 (C2′), 58.48 (Cα′), 100.91 (C2‴), 101.09 (C2), 108.06 (C4), 108.14 (C7‴ or C7), 108.36 (C7 or C7‴), 110.18 (C4‴), 119.79 (C6), 123.82 (C6‴), 128.34 (C5‴), 132.05 (C5), 146.32 (C7a), 147.05 (C7a‴), 147.26 (C3a‴), 147.72 (C3a), 169.78 (C=O). (Signals of only the predominant conformer were assigned.) Positive-ion FAB-MS m/z: 633 (M+H+). HR-FAB-MS m/z: 633.2918 (Calcd for C34H41N4O8: 633.2924). Anal. Calcd for C34H40N4O8·2H2O: C, 61.07; H, 6.63 ;N, 8.38. Found: C, 61.01; H, 6.76; N, 8.13.
N,N',N″-(Nitrilotri-2,1-ethanediyl)tris[N-(pyridin-4-ylmethyl)acetamide] (5e) (Entry 11)Compound 5e was prepared by acylation of compound 3e with Ac2O (4) under the conditions shown in Table S2. Separation of the products by flash chromatography (CH2Cl2 : 95% EtOH : 28% NH3=90 : 9.5 : 0.5→85 : 14 : 1) gave 5e (2.51 g, 61%) as a pale yellow sticky oil.
5e: IR (KBr) cm−1: 1636 (C=O), 796 (CH of pyridine). 1H-NMR (CDCl3) δ: 2.02, 2.04*, 2.07, 2.17, 2.21 (9H, s, CH3), 2.50–2.70 (6H, m, H2′), 3.18–3.45 (6H, m, H1′), 4.45, 4.50*, 4.55, 4.56, 4.58 (6H, s, Hα), 7.05–7.17 (6H, m, H3, 5), 8.52, 8.60* (6H, t, J=6.1 Hz, H2, 6). (The signals of the predominant conformer were asterisked.) 13C-NMR (CDCl3) δ: 21.46 (CH3), 44.55 (C1′), 51.59 (C2′), 51.78 (Cα), 121.15 (C3, 5), 145.92 (C4), 150.41 (C2, 6), 170.94 (C=O). (Signals of only the predominant conformer were assigned.) Positive-ion FAB-MS m/z: 546 (M+H+). HR-FAB-MS m/z: 546.3194 (Calcd for C30H40N7O3: 546.3193). Anal. Calcd for C30H39N7O3·0.5H2O: C, 64.96; H, 7.27; N, 17.68. Found: C, 65.00; H, 7.20; N, 17.46.
General Procedure for the Preparation of C3-Symmetrical Urea-Type TAEA Derivatives (7, 8): Example: Synthesis of N,N″,N-(Nitrilotri-2,1-ethanediyl)tris[N′-(3,4-dimethoxyphenyl)thiourea] (8l) (Entry 14)To a solution of 3,4-dimethoxyphenyl isothiocyanate (6l, 2.93 g, 15.0 mmol) in CH2Cl2 (20 mL) was added TAEA (1, 731 mg, 5.00 mmol) at r.t. After stirring for 1 h, the resulting white solid was filtrated to obtain crude compound 8l (3.35 g, 91%). Recrystallization from MeOH gave analytically pure compound 8l as a white solid.
8l: mp 161–163°C (from MeOH). IR (KBr) cm−1: 1513 (C=S), 1258, 1235, 1134, 1027 (C–O). 1H-NMR (CDCl3) δ: 2.67 (6H, t, J=6.9 Hz, H2′), 3.48–3.55 (6H, m, H1′), 3.72 (9H, s, –OCH3), 3.73 (9H, s, –OCH3), 6.79 (3H, dd, J=8.5, 2.1 Hz, H6), 6.90 (3H, d, J=8.5 Hz, H5), 6.96 (3H, d, J=2.1 Hz, H2), 7.36 (3H, br s, Hβ), 9.37 (3H, br s, Hα). 13C-NMR (CDCl3) δ: 41.97 (C1′), 52.19 (C2′), 55.45 (–OCH3), 55.70 (–OCH3), 109.23 (C2), 111.97 (C5), 116.28 (C6), 131.62 (C1), 146.27 (C4), 148.64 (C3), 180.28 (C=S). Positive-ion FAB-MS m/z: 732 (M+H+). HR-FAB-MS m/z: 723.2682 (Calcd for C33H46N7O6S3: 732.2672). Anal. Calcd for C33H45N7O6S3·0.8H2O: C, 53.10; H, 6.29 ; N, 13.14. Found: C, 53.08; H, 6.09; N, 13.22.
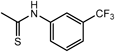
N,N″,N″″-(Nitrilotri-2,1-ethanediyl)tris[N-(3,4-dimethoxybenzyl)-N′-[3-(trifluoromethyl)phenyl]urea] (7cm) (Entry 12)Compound 7cm was prepared from 3c (282 mg, 0.473 mmol) and 3-(trifluoromethyl)phenyl isocyanate (6m, 266 mg, 1.41 mmol) under the conditions shown in Table S3. Separation of the product by flash chromatography (CH2Cl2 : 95% EtOH : 28% NH3=97 : 2.7 : 0.3→95 : 4.7 : 0.3) gave 7cm (242 mg, 44%) as a colorless oil.
7cm: 1H-NMR (CDCl3) δ: 1.64 (3H, s, NH), 2.79 (6H, br t, J=6.6 Hz, H2′), 3.55 (6H, br s, H1′), 3.82 (9H, s, MeO on C3), 3.84 (9H, s, MeO on C4), 4.74 (6H, s, Hα), 6.75–6.80 (9H, m, H2, 5, 6), 7.19 (3H, br d, J=7.5 Hz, H4″), 7.23 (3H, t, J=7.5 Hz, H5″), 7.36 (3H, br d, J=7.5 Hz, H6″), 7.58 (3H, br s, H2″). 13C-NMR (CDCl3) δ: 45.44 (C1′), 51.03 (Cα), 52.68 (C2′), 55.89 (CH3O), 55.92 (CH3O), 110.24 (C2), 111.31 (C5), 116.40 (q, J=4.3 Hz, C2″), 119.28 (q, J=4.3 Hz, C4″), 122.83 (C6″), 123.89 (q, J=271.6 Hz, CF3), 129.07 (C1), 129.19 (C5″), 131.02 (q, J=31.8 Hz, C3″), 139.72 (C1″), 148.92 (C4), 149.66 (C3), 155.87 (C=O). Positive-ion FAB-MS m/z: 1158 (M+H+). HR-FAB-MS m/z: 1158.4369 (Calcd for C57H61F9N7O9: 1158.4387).
N,N″,N″″-(Nitrilotri-2,1-ethanediyl)tris[N-(pyridin-4-ylmethyl)-N′-[4-(trifluoromethyl)phenyl]thiourea] (7ep) (Entry 13)Compound 7ep was prepared from 3e (242 mg, 0.577 mmol) and 4-(trifluoromethyl)phenyl isothiocyanate (6p, 352 mg, 1.73 mmol) under the conditions shown in Table S3. After evaporation of the solvent, recrystallization of the resulting mixture from 2-PrOH gave 7ep (296 mg, 50%) as a white solid.
7ep: mp 122–127°C (from 2-PrOH). 1H-NMR (DMSO-d6) δ: 2.95 (6H, t, J=6.7 Hz, H2′), 3.88 (6H, br s, H1′), 5.10 (6H, br s, Hα), 7.18 (6H, d, J=5.6 Hz, H3, 5), 7.48 (6H, d, J=8.4 Hz, H2″, 6″), 7.59 (6H, d, J=8.4 Hz, H3″, 5″), 8.50 (6H, d, J=5.6 Hz, H2, 6), 9.84 (3H, br s, NH). 13C-NMR (DMSO-d6) δ: 49.65 (C1′), 51.25 (C2′), 53.10 (Cα), 121.69 (C3, 5), 124.40 (q, J=32.1 Hz, C4″), 124.74 (q, J=271.1 Hz, CF3), 124.94 (q, J=4.1 Hz, C3″, 5″), 125.23 (C2″, 6″), 144.35 (C1″), 145.99 (C4), 149.56 (C2, 6), 182.21 (C=S). Positive-ion FAB-MS m/z: 1029 (M+H+). HR-FAB-MS m/z: 1029.2927 (Calcd for C48H46F9N10S3: 1029.2925).
N,N″,N-(Nitrilotri-2,1-ethanediyl)tris[N′-[3-(trifluoromethyl)phenyl]urea] (8m) (Entry 15)Compound 8m was prepared from 1 (292 mg, 2.00 mmol) and 3-(trifluoromethyl)phenyl isocyanate (6m, 1.12 g, 6.00 mmol) under the conditions shown in Table S3. After filtration of the resulting precipitates, recrystallization from propionitrile (EtCN) gave 8m (1.19 g, 84%) as colorless crystals.
8m: mp 187–190°C (from EtCN). IR (KBr) cm−1: 3334 (NH), 1645 (CONH), 1340 (CF3). 1H-NMR (DMSO-d6) δ: 2.63 (6H, t, J=6.7 Hz, H2′), 3.17–3.25 (6H, m, H1′), 6.25 (3H, t, J=5.5 Hz, NHβ), 7.20 (3H, dd, J=7.6, 0.9 Hz, H4), 7.41 (3H, t, J=7.9 Hz, H5), 7.49 (3H, d, J=8.5, H6), 7.93 (3H, s, H2), 8.87 (3H, s, NHα). 13C-NMR (DMSO-d6) δ: 37.53 (C1′), 53.72 (C2′), 113.54 (q, J=4.1 Hz, C2), 117.08 (q, J=4.1 Hz, C4), 121.06 (C6), 124.16 (q, J=272.1 Hz, CF3), 129.34 (q, J=31.0, C3), 129.52 (C5), 141.24 (C1), 155.03 (C=O). Positive-ion FAB-MS m/z: 708 (M+H+). HR-FAB-MS m/z: 708.2352 (Calcd for C30H31F9N7O3: 708.2345). Anal. Calcd for C30H30F9N7O3: C, 50.92; H, 4.27; N, 13.86. Found: C, 50.84; H, 4.07; N, 13.89.
N,N″,N-(Nitrilotri-2,1-ethanediyl)tris[N′-[4-(trifluoromethyl)phenyl]urea (8n) (Entry 16)Compound 8n was prepared from 1 (292 mg, 2.00 mmol) and 4-(trifluoromethyl)phenyl isocyanate (6n, 1.12 g, 6.00 mmol) under the conditions shown in Table S3. After filtration of the resulting precipitates, recrystallization from EtOH gave 8n (1.24 g, 88%) as colorless crystals.
8n: mp 237–238°C (from EtOH). IR (KBr) cm−1: 1649 (CONH), 1329 (CF3). 1H-NMR (DMSO-d6) δ: 2.63 (6H, t, J=6.4 Hz, H2′), 3.22 (6H, dt, J=6.4, 6.1 Hz, H1′), 6.28 (3H, t, J=5.5 Hz, NHβ), 7.52 (6H, d, J=8.9 Hz, H3, 5), 7.56 (6H, d, J=8.9 Hz, H2, 6), 8.91 (3H, s, NHα). 13C-NMR (DMSO-d6) δ: 37.52 (C1′), 53.65 (C2′), 117.20 (C2, 6), 120.89 (q, J=32.1 Hz, C4), 124.54 (q, J=271.0 Hz, CF3), 125.74 (q, J=4.1 Hz, C3, 5), 144.09 (C1), 154.84 (C=O). Positive-ion FAB-MS m/z: 708 (M+H+). HR-FAB-MS m/z: 708.2352 (Calcd for C30H31F9N7O3: 708.2345). Anal. Calcd for C30H30F9N7O3: C, 50.92; H, 4.27; N, 13.86. Found: C, 50.88; H, 4.20; N, 13.92.
N,N″,N-(Nitrilotri-2,1-ethanediyl)tris[N′-[4-(trifluoromethyl)phenyl]-thiourea] (8p) (Entry 17)Compound 8p was prepared from 1 (292 mg, 2.00 mmol) and 4-(trifluoromethyl)phenyl isothiocyanate (6p, 1.12 g, 6.00 mmol) under the conditions shown in Table S3. Filtration of the resulting precipitates gave the crude product 8p (1.12 g, 74%). Recrystallization from 2-PrOH gave analytically pure 8p as colorless crystals.
8p: mp 198–200°C (from 2-PrOH). IR (KBr) cm−1: 3275 (NH), 1548, 1542 (S=C–N), 1329 (CF3), 1111, 1067 (C=S). 1H-NMR (DNSO-d6) δ: 2.80 (6H, t, J=6.7 Hz, H2′), 3.57–3.90 (6H, m, H1′), 7.62 (6H, d, J=8.4 Hz, H3, 5), 7.70 (6H, d, J=8.4 Hz, H2, 6), 7.93 (3H, br s, NHβ), 9.88 (3H, s, NHα). 13C-NMR (DMSO-d6) δ: 41.81 (C1′), 51.86 (C2′), 121.77 (C2, 6), 123.34 (q, J=32.1 Hz, C4), 124.28 (q, J=271.0 Hz, CF3), 125.50 (q, J=3.1 Hz, C3, 5), 143.19 (C1), 180.22 (C=S). Positive-ion FAB-MS m/z: 756 (M+H+). HR-FAB-MS m/z: 756.1658 (Calcd for C30H31F9N7S3: 756.1659). Anal. Calcd for C30H30F9N7S3·2-PrOH : C, 48.58; H, 4.69; N, 12.02. Found: C, 48.54; H, 4.59; N, 11.96.
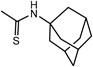
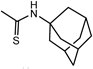
General Procedure for the Preparation of CS-Symmetrical Amine-Urea-Type TAEA Derivatives (12, 13): Example: Synthesis of N-[2-[Bis[2-[(1,3-benzodioxol-5-ylmethyl)amino]ethyl]amino]ethyl]-N′-(tricyclo[3.3.1.13,7]dec-1-yl)thiourea (12k) (Entry 18) (Step 1)To a solution of TAEA (1, 1.46 g, 10.0 mmol) in CH2Cl2 (160 mL) was added 1-adamantyl isothiocyanate (6k, 2.93 g, 15.0 mmol) in CH2Cl2 (20 mL) and the mixture was stirred for 1 h at r.t. Evaporation of the solvent gave a pale yellow oil.
(Step 2)To a solution of the resulting oil in MeOH (5 mL) was added a solution of piperonal (2d, 3.00 g, 20.0 mmol) in MeOH (10 mL) at r.t. under an argon atmosphere and the mixture was stirred for 20 h.
(Step 3)To the resulting yellow solution was added MeOH (80 mL) and NaBH4 (2.04 g, 54.0 mmol) at 0°C under an argon atmosphere with stirring for 4 h, and then stirring was continued at r.t. for 15 h. After evaporation of the solvent, aqueous ammonium acetate (NH4OAc, 10%) was added to the resulting white solid and the mixture was extracted with CHCl3 (3×200 mL). The separated organic layer was dried over MgSO4 and filtrated, and evaporation of the solvent gave a pale yellow oil. Separation of the products by flash chromatography (CH2Cl2 : 95% EtOH : 28% NH3=950 : 47 : 3→73 : 25 : 2) gave 16k (403 mg, 17%) as a white solid, 13k (365 mg, 6%) as a white solid, and 12k (2.03 g, 33%) as a colorless semisolid.
12k: mp 46–52°C. IR (KBr) cm−1: 3275 (NH of sec-amine), 1489 (C=S), 1247, 1038, 929 (C–O). 1H-NMR (CDCl3) δ: 1.58–1.67 (6H, m, Ha), 1.99 (6H, br s, Hb), 2.02 (3H, br s, Hc), 2.35 (3H, br s, NH), 2.61 (4H, br t, J=6.1 Hz, H2′), 2.66–2.71 (6H, m, H1′, 2″), 3.67 (4H, s, Hα), 5.92 (4H, s, H2), 6.09 (1H, br s, NH), 6.75 (4H, br s, H4, 6), 6.81 (2H, s, H7). 13C-NMR (CDCl3) δ: 29.44 (Cc), 36.11 (Ca), 42.03 (Cb), 43.06 (C1″), 46.63 (C1′), 53.43, 53.55, 53.62, 53.72 (C2′, 2″, α, d), 100.91 (C2), 108.14 (C4), 108.67 (C7), 121.37 (C6), 133.34 (C5), 146.72 (C3a), 147.78 (C7a), 180.73 (C=S). Positive-ion FAB-MS m/z: 608 (M+H+). HR-FAB-MS m/z: 608.3273 (Calcd for C33H46N5O4S: 608.3271). Anal. Calcd for C33H45N5O4S·0.5H2O: C, 64.26; H, 7.52; N, 11.35. Found: C, 64.32; H, 7.64; N, 11.25.
16k: 1H-NMR (CDCl3) δ: 1.69 (6H, br s, Ha), 2.13 (3H, br s, Hc), 2.15 (6H, br s, Hb), 2.70 (2H, dt, J=5.5, 0.3 Hz, H5), 3.62–3.68 (2H, m, H4), 6.49 (0.5H, br s, NH), 7.28 (0.5H, br s, NH). 13C-NMR (CDCl3) δ: 29.54 (Cc), 36.19 (Ca), 42.11 (Cb), 43.01 (C4), 53.53 (C5), 54.25 (Cd), 180.49 (C=S). Positive-ion FAB-MS m/z: 237 (32, M+H+). HR-FAB-MS m/z: 237.1417 (Calcd for C13H21N2S: 237.1425).
N,N″-[[[2-[(1,3-Benzodioxol-5-ylmethyl)amino]ethyl]imino]di-2,1-ethanediyl]bis[N′-(tricyclo[3.3.1.13,7]dec-1-yl)thiourea] (13k) (Entry 19)Compound 13k was prepared from 1 (1.46 g, 10.0 mmol) with 6k (3.87 g, 20.0 mmol) and 2d (1.50 g, 10.0 mmol) under the conditions shown in Table S4. Separation of the products by flash chromatography (CH2Cl2 : 95% EtOH : 28% NH3=95 : 4.7 : 0.3→93 : 6.6 : 0.4) gave 13k (2.12 g, 32%) as a white solid and 12k (0.903 g, 15%) as a pale yellow oil.
13k: mp 48–50°C. IR (KBr) cm−1: 3276 (NH of sec-amine), 1539 (C=S), 1247, 1038, 933 (C–O). 1H-NMR (CDCl3) δ: 1.67 (12H, br s, Ha), 2.09 (19H, br s, Hb, Hc, NH), 2.63–2.68 (2H, m, H2′), 2.68–2.74 (6H, m, H1′, 2″), 3.56–3.64 (4H, m, H1″), 3.72 (2H, s, Hα), 5.93 (2H, s, H2), 6.08 (2H, br s, NH), 6.54 (2H, br s, NH), 6.73–6.79 (2H, m, H4, 6), 6.84 (1H, d, J=1.2 Hz, H7). 13C-NMR (CDCl3) δ: 29.51 (Cc), 36.16 (Ca), 42.12 (Cb), 42.95 (C1″), 46.72 (C1′), 53.11 (C2″), 53.62 (Cα), 53.80 (C2′), 54.07 (Cd), 100.88 (C2), 108.16 (C4), 108.73 (C7), 121.41 (C6), 133.61 (C5), 146.65 (C3a), 147.74 (C7a), 180.77 (C=S). Positive-ion FAB-MS m/z: 667 (5, M+H+). HR-FAB-MS m/z: 667.3824 (Calcd for C36H55N6O2S2: 667.3828). (YO2-6) Anal. Calcd for C36H54N6O2S2·0.5H2O: C, 63.96; H, 8.20; N, 12.43. Found: C, 63.99; H, 8.19; N, 12.16.
N-[2-[Bis[2-[(1,3-benzodioxol-5-ylmethyl)amino]ethyl]amino]ethyl]-N′-(3,4-dimethoxyphenyl)thiourea (12l) and 1-(3,4-Dimethoxyphenyl)-2-imidazolidinethione (16l) (Entry 20)Compound 12l was prepared from 1 (1.46 g, 10.0 mmol) with 6l (1.95 g, 10.0 mmol) and 2d (3.00 g, 20.0 mmol) under the conditions shown in Table S4. Separation of the products by flash chromatography (CH2Cl2 : 95% EtOH : 28% NH3=93 : 6.6 : 0.4→85 : 14 : 1) gave 16l (trace) as a white solid, 13l (1.50 g, 22%) as a white solid, and 12l (2.77 g, 45%) as a white oil.
12l: IR (KBr) cm−1: 3290 (NH of sec-amine), 1513 (C=S), 1242, 1034, 928 (C–O). 1H-NMR (CDCl3) δ: 2.76 (6H, t, J=5.8 Hz, H2″, 2′), 2.82 (4H, t, J=5.8 Hz, H1′), 3.75 (2H, t, J=5.8 Hz, H1″), 3.82 (3H, s, OCH3 on C4‴), 3.82 (2H, s, Hα), 3.83 (3H, s, OCH3 on C3‴), 4.48 (3H, br s, NH), 5.92 (4H, s, H2), 6.73 (2H, d, J=7.9 Hz, H7), 6.76 (1H, d, J=8.5 Hz, H5‴), 6.85 (2H, dd, J=7.9, 1.5 Hz, H6), 6.91 (2H, d, J=1.5 Hz, H4), 6.93 (1H, dd, J=8.5, 2.4 Hz, H6‴), 7.26 (1H, br s, H2‴), 9.06 (1H, br s, NH). 13C-NMR (CDCl3) δ: 42.12 (C1″), 45.25 (C1′), 51.92 (Cα), 52.28 (C2′), 54.39 (C2″), 55.99, 56.08 (OCH3), 101.31 (C2), 108.55 (C7), 109.10 (C2‴), 109.60 (C4), 111.19 (C5‴), 116.53 (C6‴), 123.14 (C6), 127.58 (C5), 132.14 (C1‴), 146.79 (C3‴), 147.93 (C7a), 148.14 (C3a), 148.90 (C4‴), 181.62 (C=S). Positive-ion FAB-MS m/z: 610 (M+H+). HR-FAB-MS m/z: 610.2714 (Calcd for C31H40N5O6S: 610.2699). Anal. Calcd for C31H39N5O6S·0.9H2O: C, 59.48; H, 6.57; N, 11.19. Found: C, 59.51; H, 6.50; N, 11.09.
16l: 13C-NMR (CDCl3) δ: 42.98 (C4), 53.24 (C5), 56.02, 56.09 (OCH3), 109.47 (C2′), 111.70 (C5′), 117.53 (C6′), 130.34 (C1′), 147.69 (C4′), 149.41 (C3′), 181.30 (C=S). (Signals of only the predominant conformer were assigned.) Positive-ion FAB-MS m/z: 239 (M+H+). HR-FAB-MS m/z: 239.0850 (Calcd for C11H15N2O2S: 239.0854).
N,N″-[[[2-[(1,3-Benzodioxol-5-ylmethyl)amino]ethyl]imino]di-2,1-ethanediyl]bis[N′-(3,4-dimethoxyphenyl)thiourea] (13l) (Entry 21)Compound 13l was prepared from 1 (731 mg, 5.00 mmol) with 6l (1.95 g, 10.0 mmol) and 2d (751 mg, 5.00 mmol) under the conditions shown in Table S4. Separation of the products by flash chromatography (CH2Cl2: 95% EtOH : 28% NH3=95 : 4.7 : 0.3→93 : 6.6 : 0.4) gave 8l (1.11 g, 30%) as a white solid and 13l (1.15 g, 34%) as a colorless semisolid.
13l: mp 65–69°C. IR (KBr) cm−1: 3264 (NH of sec-amine), 1512 (C=S), 1236, 1026, 927 (C–O). 1H-NMR (CDCl3) δ: 1.81 (2H, br s, NH), 2.59 (4H, br s, H1′, 2′), 2.66 (4H, t, J=6.1 Hz, H2″), 3.56 (2H, s, Hα), 3.63 (2H, br s, H1″), 3.82 (6H, s, CH3O on C3‴), 3.84 (6H, s, CH3O on C4‴), 5.87 (2H, s, H2), 6.66–6.71 (2H, m, H7, 6), ca. 6.7 (1H, br s, NH), 6.73 (1H, br s, H4), 6.76 (2H, dd, J=8.2, 2.1 Hz, H6‴), 6.82 (2H, d, J=8.2 Hz, H5‴), 6.83 (2H, br s, H2‴), 7.88 (2H, br s, NH). 13C-NMR (CDCl3) δ: 43.48 (C1″), 46.77 (C1′), 53.36 (Cα), 53.43 (C2′ or 2″), 53.62 (C2″ or 2′), 56.09, 56.12 (OCH3), 100.89 (C2), 108.11 (C7 or 4), 108.59 (C4 or 7), 109.83 (C2‴), 111.65 (C5‴), 118.04 (C6‴), 121.25 (C6), 129.69 (C1‴), 133.50 (C5), 146.61 (C3a or 7a), 147.72 (C4‴), 148.14 (C7a or 3a), 149.67 (C3‴), 181.27 (C=S). Positive-ion FAB-MS m/z: 671 (M+H+). HR-FAB-MS m/z: 671.2684 (Calcd for C32H43N6O6S2: 671.2686). Anal. Calcd for C32H42N6O6S2·1.2H2O: C, 55.50; H, 6.46; N, 12.14. Found: C, 55.54; H, 6.24; N, 11.86.
N-[2-[Bis[2-[(1,3-benzodioxol-5-ylmethyl)amino]ethyl]amino]ethyl]-N′-[3-(trifluoromethyl)phenyl]thiourea (12o), N,N′-[[[2-[(1,3-Benzodioxol-5-ylmethyl)amino]ethyl]imino]di-2,1-ethanediyl]bis[N′-[3-(trifluoromethyl)phenyl]thiourea] (13o) and 1-[3-(Trifluoromethyl)phenyl]-2-imidazolidinethione (16o) (Entry 22)Compounds 12o and 13o were prepared from 1 (1.46 g, 10.0 mmol) with 6o (2.03 g, 10.0 mmol) and 2d (3.00 g, 20.0 mmol) under the conditions shown in Table S4. Separation of the products by flash chromatography (CH2Cl2 : 95% EtOH : 28% NH3=93 : 6.6 : 0.4→85 : 14 : 1) gave 16o (466 mg, 19%) as a pale yellow semi-solid, 13o (1.51 g, 22%) as a pale yellow semi-solid, and 12o (3.42 g, 55%) as a pale yellow oil.
12o: IR (KBr) cm−1: 3260 (NH of sec-amine), 1490 (C=S), 1332 (ArCF3), 1249, 1038, 930 (C–O). 1H-NMR (CDCl3) δ: ca. 2.3 (4H, br s, NH), 2.58 (4H, t, J=5.5 Hz, H2′), 2.65–2.70 (6H, m, H1′, 2″), 3.60 (4H, s, Hα), 3.60 (2H, br s, H1″), 5.87 (4H, s, H2), 6.67–6.71 (4H, m, H6, 7), 6.74 (2H, s, H4), 7.34–7.37 (2H, m, H4‴, 5‴), 7.56 (2H, br s, H2‴, 6‴). 13C-NMR (CDCl3) δ: 43.24 (C1″), 46.66 (C1′), 52.57 (C2″), 53.46 (Cα), 53.95 (C2′), 100.95 (C2), 108.19 (C7), 108.57 (C4), 120.36 (q, J=4.1 Hz, C2‴), 121.30 (C6), 121.49 (q, J=4.1 Hz, C4‴), 125.96 (q, J=272.1 Hz, CF3), 127.05 (C6‴), 129.22 (C5‴), 131.21 (q, J=33.1 Hz, C3‴), 133.20 (C5), 139.29 (C1‴), 146.77 (C3a), 147.83 (C7a), 181.05 (C=S). Positive-ion FAB-MS m/z: 135 (100), 247 (14), 618 (6, M+H+). HR-FAB-MS m/z: 618.2363 (Calcd for C30H35F3N5O4S: 618.2362). Anal. Calcd for C30H34F3N5O4S·0.6H2O: C, 57.33; H, 5.65; N, 11.14. Found: C, 57.35; H, 5.65; N, 10.94.
13o: IR (KBr) cm−1: 3256 (NH of sec-amine), 1490 (C=S), 1332 (ArCF3), 1251, 1038, 930 (C–O). 1H-NMR (CDCl3) δ: 1.23 (3H, t, J=7.0 Hz, CH3 of EtOH), ca. 2.15 (3H, br s, NH and/or OH), 2.66–2.71 (2H, m, H2′), ca. 2.7 (1H, br s, NH or OH), 2.71–2.78 (6H, m, H1′, 2″), 3.57 (2H, s, Hα), 3.69 (4H, br s, H1″), 3.70 (2H, q, J=7.0 Hz, CH2 of EtOH), ca. 3.7 (1H, br s, NH), 5.78 (2H, s, H2), 6.57–6.68 (3H, m, H6, 7, 4), ca. 6.65 (1H, br s, NH), 7.22–7.31 (4H, m, H4‴, 5‴), 7.36 (2H, br d, J=7.6 Hz, H6‴), 7.59 (2H, s, H2‴). 13C-NMR (CDCl3) δ: 18.40 (CH3 of EtOH), 43.36 (C1″), 46.82 (C1′), 53.29 (C2′), 53.59 (Cα), 53.66 (C2″), 58.34 (CH2 of EtOH), 100.99 (C2), 108.33 (C7), 108.61 (C4), 120.45 (q, J=4.1 Hz, C2‴), 121.56 (C6), 121.86 (q, J=4.1 Hz, C4‴), 123.68 (q, J=272.1 Hz, CF3), 127.07 (C6‴), 129.25 (C5‴), 131.07 (q, J=30.0 Hz, C3‴), 132.27 (C5), 138.90 (C1‴), 146.93 (C3a), 147.85 (C7a), 181.20 (C=S). Positive-ion FAB-MS m/z: 135 (100), 247 (61), 687 (22, M+H+). HR-FAB-MS m/z: 687.2006 (Calcd for C30H33F6N6O2S2: 687.2011). Anal. Calcd for C30H32F6N6O2S2·EtOH: C, 52.45; H, 5.23; N, 11.47. Found: C, 52.44; H, 5.00; N, 11.46.
16o: Positive-ion FAB-MS m/z: 135 (57), 247 (100, M+H+). HR-FAB-MS m/z: 247.0515 (Calcd for C10H10F3N2S: 247.0517). 1H-NMR (CDCl3) δ: 2.70 (2H, t, J=4.8 Hz, H5), 3.80 (2H, br d, J=4.8 Hz, H4), 7.20–7.24 (1H, m, H5′), 7.26 (1H, d, J=6.8 Hz, H4′), 7.50 (1H, br d, J=6.9 Hz, H6′), 7.91 (1H, br s, NH), 7.97 (1H, br s, H2′). 13C-NMR (CDCl3) δ: 41.97 (C4), 52.57 (C5), 119.65 (br s, C2′), 121.11 (C4′), 123.79 (q, J=273.1 Hz, CF3), 126.27 (C6′), 129.11 (C5′), 130.66 (q, J=33.2 Hz, C3′), 139.58 (C1′), 180.88 (C=S).
General Procedure for the Preparation of CS-Symmetrical Amide-Urea-Type TAEA Derivatives (14, 15): Example: Synthesis of N-(1,3-Benzodioxol-5-ylmethyl)-N-[2-[Bis[2-[[thioxo(tricyclo[3.3.1.13,7]dec-1-ylamino)methyl]amino]ethyl]amino]ethyl]acetamide (15k) (Entry 24) (Step 1)To a pale yellow suspension of compound 13k (1.88 g, 2.82 mmol) in tetrahydrofuran (THF) (5 mL) was added Ac2O (4, 288 mg, 2.82 mmol) and the mixture was stirred for 30 min at r.t. The crude precipitated solid was obtained as a white substance 15k (1.83 g, 92%). Recrystallization from THF gave an analytically pure sample as a white powder.
15k: mp 188–192°C (from THF). IR (KBr) cm−1: 1627 (C=O), 1540 (C=S), 1357, 1238, 1038 (C–O). 1H-NMR (CDCl3) δ: 1.68 (12H, br s, Ha), 2.11 (6H, br s, Hc), 2.14 (12H, br s, Hb), 2.19 (3H, s, CH3), 2.63–2.68 (6H, m, H2′, 2″), 3.43 (2H, t, J=5.5 Hz, H1′), 3.55 (4H, q, J=5.2 Hz, H1″), 4.53 (2H, br s, Hα), 5.96 (2H, s, H2), 6.32 (2H, br s, NH), 6.62 (2H, br s, NH), 6.63 (1H, d, J=7.9 Hz, H6), 6.64 (1H, br s, H4), 6.7–6.8 (1H, d, J=7.9 Hz, H7). 13C-NMR (CDCl3) δ: 22.26 (CH3), 29.60 (Cc), 36.31 (Ca), 42.17 (Cb), 42.86 (C1″), 43.89 (C1′), 51.53 (Cα), 52.53 (C2′), 53.19 (C2″), 53.94 (Cd), 101.27 (C2), 106.75 (C4), 108.65 (C7), 119.63 (C6), 129.72 (C5), 147.30 (C3a), 148.42 (C7a), 172.52 (C=O), 181.19 (C=S). Positive-ion FAB-MS m/z: 709 (M+H+). HR-FAB-MS m/z: 709.3948 (Calcd for C38H57N6O3S2: 709.3934). Anal. Calcd for C38H56N6O3S2·0.4H2O: C, 63.72; H, 7.99; N, 11.73. Found: C, 63.69; H, 8.13; N, 11.47.
N,N′-[[[2-[[Thioxo(tricyclo[3.3.1.13,7]dec-1-ylamino)methyl]amino]ethyl]imino]di-2,1-ethanediyl]bis[N-(1,3-benzodioxol-5-ylmethyl)acetamide] (14k) (Entry 23)Compound 14k was prepared from 12k (1.22 g, 2.00 mmol) and Ac2O (4, 408 mg, 4.00 mmol) under the conditions shown in Table S5. Separation of the product by flash chromatography (EtOAc : EtOH=99 : 1) gave 14k (934 mg, 68%) as a white solid.
14k: mp 66–68°C. IR (KBr) cm−1: 3324 (NH), 1637 (C=O), 1490 (C=S), 1248, 1038, 924 (C–O). 1H-NMR (CDCl3) δ: 1.66 (6H, br s, Ha), 2.06 (3H, br s, Hc), 2.12*, 2.16 (6H, s, CH3), 2.08, 2.20* (6H, br s, Hb), 2.6–2.55 (4H, m, H2′), 2.6–2.7 (2H, m, H2″), 3.2–3.25, 3.3–3.4* (4H, m, H1′), 3.4–3.5, 3.5–3.6* (2H, m, H1″), 4.36, 4.45*, 4.48 (4H, s, Hα), 5.92, 5.96*, 5.97 (4H, s, H2), 6.34 (1H, br s, NH), 6.6–6.7 (4H, m, H4, 6), 6.7–6.8 (2H, m, H7), 7.18 (1H, br s, NH). (The signals of the predominant conformer were asterisked.) 13C-NMR (CDCl3) δ: 21.84 (CH3), 29.51 (Cc), 36.30 (Ca), 41.88 (Cb), 42.41 (C1″), 43.38 (C1′), 51.34 (Cα), 51.99 (C2′), 52.69 (C2″), 53.27 (Cd), 101.09 (C2), 106.66 (C4), 108.50 (C7), 119.45 (C6), 130.07 (C5), 147.05 (C3a), 148.23 (C7a), 171.50 (C=O), 180.82 (C=S). (Signals of only the predominant conformer were assigned.) Positive-ion FAB-MS m/z: 692 (M+H+). HR-FAB-MS m/z: 692.3488 (Calcd for C37H50N5O6S: 692.3482). Anal. Calcd for C37H49N5O6S·0.9H2O: C, 62.76; H, 7.23; N, 9.89. Found: C, 62.87; H, 7.33; N, 9.64.
N,N′-[[[2-[[Thioxo(3,4-dimethoxyphenylamino)methyl]amino]ethyl]imino]di-2,1-ethanediyl]bis[N-(1,3-benzodioxol-5-ylmethyl)acetamide] (14l) (Entry 25)Compound 14l was prepared from 12l (1.53 g, 2.51 mmol) and 4 (0.512 g, 5.02 mmol) under the conditions shown in Table S5. Separation of the product by centrifugal chromatography (CH2Cl2 : 95% EtOH : 28% NH3=95 : 4.7 : 0.3→93 : 6.6 : 0.4) gave 14l (1.39 g, 80%) as a white solid.
14l: mp 54–58°C. IR (KBr) cm−1: 1639 (C=O), 1512, 1490 (C=S), 1237, 1036, 923 (C–O). 1H-NMR (CDCl3) δ: 2.03, 2.08, 2.10*, 2.11 (6H, s, CH3CO), 2.45–2.6 (4H, m, H2′), 2.6–2.70 (2H, m, H2″), ca. 2.65 (1H, br s, OH), 3.05–3.15, 3.24–3.3, 3.3–3.35* (4H, m, H1′), 3.57 (2H, br s, H1″), 3.80, 3.82*, 3.83 (6H, s, OCH3), 4.33, 4.42*, 4.44 (4H, s, Hα), 5.91, 5.95*, 5.95 (4H, s, H2), 6.55–6.95, 7.17–7.23 (9H, m, H6, 4, 7, 5‴, 6‴, 2‴), 7.49, 8.30, 8.63* (2H, br s, NH). (The signals of the predominant conformer were asterisked.) 13C-NMR (CDCl3) δ: 21.64 (CH3CO), 42.46 (C1″), 43.61 (C1′), 51.53 (Cα), 51.88 (C2′), 52.74 (C2″), 55.71, 55.84 (OCH3), 100.99 (C2), 106.58 (C4), 108.32 (C7), 108.91 (C2‴), 111.19 (C5‴), 116.22 (C6‴), 119.41 (C6), 129.92 (C5), 131.31 (C1‴), 146.96 (C3a or C7a), 147.72 (C4‴), 148.09 (C7a or C3a), 148.84 (C3‴), 171.32 (C=O), 181.02 (C=S). (Signals of only the predominant conformer were assigned.) Positive-ion FAB-MS m/z: 694 (M+H+). HR-FAB-MS m/z: 694.2928 (Calcd for C35H44N5O8S: 694.2911). Anal. Calcd for C35H43N5O8S·0.5H2O; C, 59.81; H, 6.31; N, 9.96. Found: C, 59.74; H, 6.31; N, 10.04.
N-(1,3-Benzodioxol-5-ylmethyl)-N-[2-[Bis[2-[[thioxo(3,4-dimethoxyphenylamino)methyl]amino] Ethyl]amino]ethyl]acetamide (15l) (Entry 26)Compound 15l was prepared from 13l (394 mg, 587 µmol) and 4 (59.9 mg, 587 µmol) under the conditions shown in Table S5. Separation of the product by centrifugal chromatography (CH2Cl2 : EtOH=95 : 5) gave 15l (368 mg, 88%) as a white solid. An analytical sample of 15l was obtained by recrystallization from dichloromethane (CH2Cl2) as an off-white solid.
15l: mp 193–194°C (from CH2Cl2). IR (KBr) cm−1: 1616 (C=O), 1511, 1453 (C=S), 1239, 1034, 922 (C–O). 1H-NMR (DMSO-d6) δ: 1.99, 2.08* (3H, s, CH3CO), 2.54–2.66 (6H, m, H2′, 2″), 3.18–3.27 (2H, m, H1′), 3.49 (4H, br t, J=5.5 Hz, H1″), 3.71 (6H, s, CH3 on C3‴), 3.72 (6H, s, CH3 on C4‴), 4.39*, 4.41 (2H, br s, Hα), 5.70*, 6.00 (2H, s, H2), 6.64–6.98 (9H, m, H6, 7, 4, 6‴, 5‴, 2‴), 7.31 (2H, br s, NHβ), 9.39 (2H, s, NHγ). (The signals of the predominant conformer were asterisked.) 13C-NMR (DMSO-d6) δ: 21.15 (CH3CO), 41.94 (C1″), 45.93 (C1′), 47.64 (Cα), 51.71 (C2′), 52.40 (C2″), 55.44, 55.68 (OCH3), 100.76 (C2), 107.93 (C7), 108.08 (C4), 109.30 (C2‴), 111.98 (C5‴), 116.39 (C6‴), 120.96 (C6), 131.42 (C1‴ or C5), 132.12 (C5 or C1‴), 146.27 (C3a or C7a), 146.38 (C4‴), 147.29 (C7a or C3a), 148.70 (C3‴), 169.64 (C=O), 180.26 (C=S). (Signals of only the predominant conformer were assigned.) Positive-ion FAB-MS m/z: 713 (M+H+). HR-FAB-MS m/z: 713.2786 (Calcd for C34H45N6O7S2: 713.2791). Anal. Calcd for C34H44N6O7S2: C, 57.29; H, 6.22; N, 11.79. Found: C, 57.10; H, 6.17; N, 11.75.
Preparation of C3-Symmetrical TAPA Derivatives (18a, 21) 2,2′,2″-[Nitrilotris(3,1-propanediyliminomethylene)]tris[6-methoxyphenol] (18a)Compound 18a was prepared starting from the reaction of TAPA (17, 942 mg, 5.0 mmol) and 2a (3.04 g, 20.0 mmol) in a manner similar to that for the preparation of compound 3a. The reactions in both steps were conducted in EtOH instead of MeOH with the ratio of TAPA : 2a : NaBH4=1 : 4 : 8.2. Separation of the product by centrifugal chromatography (CHCl3 : 2-PrOH : 28% NH3=84 : 14 : 2) gave 18a (1.93 g, 65%) as a light-brown semisolid.
18a: mp 31–35°C. IR (KBr) cm−1: 3420 (OH), 3310 (NH), 1590, 1480 (C=C of Ar), 1235, 1075 (C–O). 1H-NMR (CDCl3) δ: 1.63 (6H, qu, J=6.7 Hz, H2′), 2.41 (6H, t, J=6.7 Hz, H3′), 2.64 (6H, t, J=6.7 Hz, H1′), 3.84 (9H, s, CH3O), 3.93 (6H, s, Hα), 6.26 (6H, br s, NH, OH), 6.61 (3H, d, J=7.6 Hz, H3), 6.71 (3H, dd, J=7.9, 7.6 Hz, H4), 6.79 (3H, d, J=7.9 Hz, H5). 13C-NMR (CDCl3) δ: 26.74 (C2′), 47.35 (C1′), 52.21 (C3′ or Cα), 52.24 (Cα or C3′), 55.83 (CH3O), 110.83 (C5), 118.48 (C4), 120.55 (C3), 122.69 (C2), 147.40 (C1), 147.96 (C6). Positive-ion FAB-MS m/z: 597 (M+H+). HR-FAB-MS m/z: 597.3644 (Calcd for C33H49N4O6: 597.3652). Anal. Calcd for C33H48N4O6·1.2 H2O: C, 64.10; H, 8.22; N, 9.06. Found: C, 64.15; H, 8.31; N, 8.91.
N,N′,N″-(Nitrilotri-3,1-propanediyl)tris[3,5-bis(trifluoromethyl)benzamide] (21)To a solution of 17 (942 mg, 5.0 mmol) and triethylamine (TEA, 1.77 g, 17.5 mmol) in CH2Cl2 (50 mL) was added a solution of 3,5-bis(trifluoromethyl)benzoyl chloride (19, 4.78 g, 17.3 mmol) in CH2Cl2 (25 mL) at 0°C under an N2 atmosphere. After stirring for 30 min, the reaction mixture was stirred at r.t. for another 1 h. The resulting mixture was poured into water (100 mL), and then the aqueous layer was extracted with CH2Cl2 (2×100 mL). The combined organic layer was washed with brine (50 mL) and dried over MgSO4, and the solvent was evaporated to give a yellow oil. Separation of the product by flash chromatography (CH2Cl2 : 95% EtOH : 28% NH3=97 : 2.7 : 0.3) and then recrystallization from MeOH gave compound 21 (2.99 g, 66%) as colorless crystals.
21: mp 205–206°C (from MeOH). IR (KBr) cm−1: 3265 (NH), 1645, 1550, 1280 (C=O), 1135 (CF3). 1H-NMR (DMSO-d6) δ: 1.71(6H, t, J=7.0 Hz, H2′), 2.49 (6H, m, H3′), 3.35 (6H, m, H1′), 8.23 (3H, s, H4), 8.41 (6H, s, H2, H6), 8.94 (3H, t, J=5.3 Hz, NH). 13C-NMR (DMSO-d6) δ: 26.39 (C2′), 38.03 (C1′), 50.93 (C3′), 123.08 (CF3, q, J=273.1 Hz), 124.58 (C4), 127.86 (C2, C6), 130.41 (C3, C5, q, J=33.1 Hz), 136.68 (C1), 163.13 (C=0). Positive-ion FAB-MS m/z: 909 (M+H+). HR-FAB-MS m/z: 909.2101 (Calcd for C36H31F18N4O3: 909.2109). Anal. Calcd for C36H30F18N4O3: C, 47.59; H, 3.33; N, 6.17. Found: C, 47.58; H, 3.34; N, 6.22.
Antiviral Activity Assay and Cytotoxicity of Synthesized Trisubstituted Tris(aminoalkyl)amine DerivativesThe anti-HSV-1 activities (EC50) of the synthesized tris(aminoalkyl)amine derivatives were assessed by using a plaque reduction assay17) and their cytotoxicity against Vero cells (CC50) was also evaluated. The results are summarized in Tables 1, 2 together with data for aciclovir.18) Log P values for all of the compounds for which biological activities were evaluated were calculated by using CAChe v.6.1.12. There were few distinct correlations between log P values and anti-HSV-1 activities (EC50 values) among the compounds listed in Tables 1, 2.