Experimental
GeneralReactions were performed under argon unless otherwise stated. Commercially available chemicals were used as purchased. Special grade and dehydrated solvents were purchased from Kanto Chemical Co., Inc. (Tokyo, Japan). The progress of the reaction was monitored by TLC using precoated silica gel 60 F254 plates (Merck, Darmstadt, Germany). Spots were visualized under UV illumination (254 nm) after immersion in 2% p-anisaldehyde and 5% H2SO4 in EtOH, followed by heating on a hot plate. Solvents were removed under reduced pressure using a standard rotary evaporator. Flash column chromatography was performed using 63–210 mesh silica gel 60 (Kanto Chemical Co., Inc.). Specific rotations were determined on a JASCO P-1030 digital polarimeter using the sodium D line (λ=589 nm) at specific temperatures and are reported as follows: [α]Dtemp., concentration (c=g/100 mL), and solvent. Fourier transform (FT)-IR spectra were measured on a JASCO FT/IR-410 infrared spectrophotometer and reported as wavenumber (cm−1) of significant peaks. 1H- and 13C-NMR spectra were recorded on a Varian Mercury plus-300-4N spectrometer, a Varian 400-MR spectrometer, a Varian 500-MR spectrometer and a Varian Unity-600 spectrometer. The 1H chemical shifts are reported in parts per million (ppm) from an internal standard of tetramethylsilane (TMS) (0.00 ppm), residual CHCl3 (7.26 ppm), or C5H5N (7.19 ppm, right peak), and the 13C chemical shifts were reported using an internal standard of CDCl3 (77.03 ppm, central peak) or C5D5N (123.50 ppm, central peak in right-side triplet). The 1H-NMR spectra are reported as follows: chemical shift, multiplicity (s=singlet, d=doublet, t=triplet, q=quartet, qui=quintet and br=broad), relative integral, coupling constants J (Hz). High-resolution (HR)-MS were recorded on a JEOL the Mstation JMS-700.
Allyl 4,6-O-Benzylidene-2-deoxy-3-O-(3-nonyldodecanoyl)-2-(2,2,2-trichloroethoxycarbonylamino)-α-D-glucopyranoside (11)To a solution of 827) (18.6 g, 39.0 mmol) in CH2Cl2 (130 mL) were added 3-nonyldodecanoic acid (16.6 g, 50.8 mmol), MS 4 Å (30.0 g), 4,4-dimethylaminopyridine (1.40 g, 11.7 mmol) and EDCI (11.2 g, 58.5 mmol) at room temperature. After the reaction mixture was stirred for 4 h at reflux, it was cooled to room temperature and then filtrated by Celite®. The filtrate was diluted with CH2Cl2 and quenched by H2O. The organic layer was separated, and the aqueous layer was extracted with CH2Cl2 three times. The combined organic layer was dried over MgSO4, and concentrated in vacuo. The residue was purified by silica gel chromatography (hexane–AcOEt=10 : 1) to give 11 as colorless syrup (28.6 g, 36.1 mmol, 93%). 11: [α]D21 +28.1 (c=1.0, CHCl3); FT-IR (neat) 3323, 3065, 3036, 3013, 2954, 2924, 2852, 1742, 1722, 1647 cm−1; 1H-NMR (300 MHz in CDCl3) δ: 0.88 (6H, m, CH3×2), 1.24 (32H, m, CH2×16), 1.80 (1H, m, β-CH), 2.23 (2H, t, J=6.6 Hz, α-CH2), 3.74 (2H, m, H-4 and H-6b), 4.00 (3H, m, H-2, H-5 and OCH2CH=CH2), 4.22 (1H, dd, J=12.9, 5.4 Hz, OCH2CH=CH2), 4.29 (1H, dd, J=10.2, 4.8 Hz, H-6a), 4.66 (1H, d, J=12.0 Hz, CH2 of Troc), 4.77 (1H, d, J=12.0 Hz, CH2 of Troc), 4.94 (1H, d, J=3.9 Hz, H-1), 5.28 (2H, m, OCH2CH=CH2), 5.41 (2H, m, H-3 and NH), 5.53 (1H, s, PhCH(CO)2), 5.89 (1H, m, OCH2CH=CH2), 7.39 (5H, m, PhCH(CO)2); 13C-NMR (75 MHz in CDCl3) δ: 14.16, 22.71, 26.39, 26.43, 29.37, 29.64, 29.67, 29.84, 29.87, 31.93, 33.40, 34.87, 38.95, 54.74, 63.03, 68.37, 69.79, 74.59, 79.26, 95.38, 97.02, 101.44, 118.55, 126.06, 128.15, 129.01, 133.03, 136.90, 154.31, 173.40; MS (FAB+) m/z 789 [M]+; HR-MS (FAB+) m/z Calcd for C40H62NO8Cl3: 789.3541. Found 789.3556.
Allyl 6-O-Benzyl-2-deoxy-3-O-(3-nonyldodecanoyl)-2-(2,2,2-trichloroethoxycarbonylamino)-α-D-glucopyranoside (12)To a solution of 11 (10.0 g, 12.6 mmol) in CH2Cl2 (85 mL) were added triethylsilane (7.30 g, 63.0 mmol) and BF3·Et2O (8.90 g, 63.0 mmol) at 0°C. After the mixture was stirred for 1 h at 0°C, it was quenched with triethylamine and concentrated in vacuo. The residue was purified by silica gel chromatography (hexane–AcOEt=10 : 1) to give 12 as colorless syrup (9.00 g, 11.3 mmol, 90%). 12: [α]D21 +38.6 (c=1.0, CHCl3); FT-IR (neat) 3437, 3339, 3086, 3065, 3031, 2927, 2855, 1950, 1864, 1743, 1649 cm−1; 1H-NMR (300 MHz in CDCl3) δ: 0.88 (6H, m, CH3×2), 1.25 (32H, m, CH2×16), 1.82 (1H, m, β-CH), 2.27 (2H, t, J=6.0 Hz, α-CH2), 2.68 (1H, d, J=3.6 Hz, OH), 3.78 (4H, m, H-4, H-5 and H-6ab), 3.98 (2H, m, H-2 and OCH2CH=CH2), 4.19 (1H, dd, J=12.6, 5.1 Hz, OCH2CH=CH2), 4.57 (1H, d, J=12.0 Hz, CH2 of Troc), 4.63 (1H, d, J=12.0 Hz, CH2 of Troc), 4.65 (1H, d, J=12.0 Hz, PhCH2), 4.74 (1H, d, J=12.0 Hz, PhCH2), 4.91 (1H, d, J=3.6 Hz, H-1), 5.21 (4H, m, H-3, NH and OCH2CH=CH2), 5.87 (1H, m, OCH2CH=CH2), 7.34 (5H, m, PhCH2); 13C-NMR (75 MHz in CDCl3) δ: 14.11, 22.66, 26.35, 26.56, 29.32, 29.59, 29.63, 29.88, 29.89, 31.88, 33.51, 33.60, 34.82, 39.00, 53.79, 68.43, 69.59, 70.24, 70.46, 73.42, 73.64, 74.54, 95.37, 96.24, 118.21, 127.59, 127.75, 128.39, 133.18, 137.70, 154.18, 174.72; MS (FAB+) m/z 814 [M+H+Na]+; HR-MS (FAB+) m/z Calcd for C40H64NO8Cl3Na: 814.3595. Found 814.3601.
Allyl 6-O-Benzyl-2-deoxy-3-O-(3-nonyldodecanoyl)-4-O-(3-oxido-1,5-dihydrobenzo[e][1,3,2]dioxaphosphepin-3-yl)-2-(2,2,2-trichloroethoxycarbonylamino)-α-D-glucopyranoside (13)To a solution of 12 (2.10 g, 2.70 mmol) in 1,2-dichloroethane (5.4 mL) were added MS 4 Å (1.50 g), N,N-diethyl-1,5-dihydro-2,3,4-benzodioxaphosphepin-3-amine (0.8 mL, 3.70 mmol) and 1H-tetrazole (476 mg, 6.80 mmol) at room temperature. After the mixture was stirred for 10 min at 0°C, m-chloroperoxybenzoic acid (contain 55%) (467 mg, 2.70 mmol) was added to the mixture at −20°C. After the mixture was stirred for 10 min at −20°C, it was filtrated by Celite®. The filtrate was diluted with AcOEt and quenched with aqueous NaHCO3 and Na2S2O3. The combined organic layer was dried over MgSO4, and concentrated in vacuo. The residue was purified by silica gel chromatography (hexane–AcOEt=4 : 1) to give 13 as colorless syrup (2.61 g, 2.67 mmol, 99%). 13: [α]D22 +29.2 (c=1.0, CHCl3); FT-IR (neat) 3437, 3331 3064, 3029, 2927, 2855, 1958, 1864, 1804, 1747, 1647 cm−1; 1H-NMR (300 MHz in CDCl3) δ: 0.87 (3H, t, J=6.6 Hz, CH3), 0.88 (3H, t, J=6.6 Hz, CH3), 1.24 (32H, m, CH2×16), 1.82 (1H, m, β-CH), 2.34 (2H, d, J=6.6 Hz, α-CH2), 3.73 (1H, dd, J=10.8, 4.8 Hz, H-6a), 3.80 (1H, dd, J=10.8, 2.1 Hz, H-6b), 4.01 (3H, m, H-2, H-5 and OCH2CH=CH2), 4.21 (1H, dd, J=12.9, 5.4 Hz, OCH2CH=CH2), 4.55 (1H, d, J=12.3 Hz, CH2 of Troc), 4.58 (1H, d, J=12.3 Hz, CH2 of Troc), 4.66 (1H, d, J=12.3 Hz, PhCH2), 4.74 (1H, m, H-4), 4.81 (1H, d, J=12.3 Hz, PhCH2), 4.95 (1H, d, J=3.9 Hz, H-1), 5.10 (4H, m, o-C6H4(CH2O)2P), 5.30 (4H, m, H-3, NH and OCH2CH=CH2), 5.88 (1H, m, OCH2CH=CH2), 7.18–7.40 (9H, m, o-C6H4(CH2O)2P and PhCH2); 13C-NMR (75 MHz in CDCl3) δ: 14.14, 22.69, 26.47, 26.61, 29.36, 29.66, 29.73, 30.02, 31.91, 33.38, 33.62, 34.33, 38.69, 54.13, 68.28, 68.59, 68.72, 69.84, 70.75, 73.62, 74.42, 74.64, 95.29, 96.06, 118.47, 127.59, 127.85, 128.33, 128.41, 128.49, 128.95, 133.02, 134.68, 137.89, 154.03, 173.86; MS (FAB+) m/z 996 [M+Na]+; HR-MS (FAB+) m/z Calcd for C48H71NO11Cl3PNa: 996.3728. Found 996.3715.
6-O-Benzyl-2-deoxy-3-O-(3-nonyldodecanoyl)-4-O-(3-oxido-1,5-dihydrobenzo[e][1,3,2]dioxaphosphepin-3-yl)-2-(2,2,2-trichloroethoxycarbonylamino)-α-D-glucopyranose (14)To a solution of 13 (2.50 g, 2.60 mmol) in THF (52 mL, degassed with Ar) was added [Ir(cod)(MePh2P)2]PF6 (60.0 mg, 0.20 mmol) at room temperature. The mixture was stirred for 1 h under bubbling H2 gas at room temperature. H2O (32.5 mL) and I2 (660 mg, 2.60 mmol) were added to the mixture at room temperature. After the mixture was stirred for 10 min at room temperature, it was diluted with AcOEt and quenched with saturated Na2S2O3 solution. The organic layer was separated and washed with saturated NaHCO3 solution and then brine. The combined organic layer was dried over MgSO4, and concentrated in vacuo. The residue was purified by silica gel chromatography (hexane–AcOEt=2 : 1) to give 14 as colorless syrup (2.00 g, 2.14 mmol, 80%). 14: [α]D22 +6.6 (c=1.0, CHCl3); FT-IR (neat) 3374, 3109, 3066, 3026, 2955, 2925, 2853, 2737, 1949, 1910, 1742, 1725 cm−1; 1H-NMR (500 MHz in CDCl3) δ: 0.87 (3H, t, J=7.0 Hz, CH3), 0.87 (3H, t, J=6.5 Hz, CH3), 1.25 (32H, m, CH2×16), 1.80 (1H, m, β-CH), 2.32 (2H, t, J=6.0 Hz, α-CH2), 3.69 (1H, dd, J=11.0, 6.0 Hz, H-6a), 3.78 (1H, dd, J=11.0, 2.0 Hz, H-6b), 3.95 (1H, dt, J=10.5, 3.5 Hz, H-2), 4.24 (1H, m, H-5), 4.55 (1H, d, J=12.0 Hz, CH2 of Troc), 4.58 (1H, d, J=12.0 Hz, CH2 of Troc), 4.60 (2H, m, H-4 and PhCH2), 4.75 (1H, d, J=12.0 Hz, PhCH2), 5.04 (4H, m, o-C6H4(CH2O)2P), 5.25 (1H, d, J=3.5 Hz, H-1), 5.39 (2H, m, H-3 and NH), 7.16–7.37 (9H, m, o-C6H4(CH2O)2P and PhCH2); 13C-NMR (75 MHz in CDCl3) δ: 14.14, 22.70, 26.46, 26.62, 29.37, 29.68, 29.75, 29.99, 30.04, 31.92, 33.38, 33.61, 34.28, 38.68, 54.33, 68.32, 68.72, 69.17, 70.40, 74.70, 91.45, 95.24, 127.85, 128.14, 128.43, 128.52, 129.00, 134.58, 134.63, 137.46, 154.06, 173.78; MS (FAB+) m/z 956 [M+Na]+; HR-MS (FAB+) m/z Calcd for C45H69NO11Cl3PNa: 956.3415. Found 956.3415.
Allyl 6-O-{6′-O-Benzyl-2′-deoxy-3′-O-(3-nonyldodecanoyl)-4′-O-(3-oxido-1,5-dihydrobenzo[e][1,3,2]dioxaphosphepin-3-yl)-2′-(2,2,2-trichloroethoxycarbonylamino)-β-D-glucopyranosyl}-2-deoxy-3-O-[(R)-3-(benzyloxy)tetradecanoyl]-2-O-[(R)-3-(benzyloxy)tetradecanoylamino]-α-D-glucopyranoside (15)To a solution of 14 (1.00 g, 1.00 mmol) in CH2Cl2 (50 mL) were added Cs2CO3 (160 mg, 0.450 mmol) and trichloroacetonitrile (1.40 g, 10.0 mmol) at room temperature. After the mixture was stirred for 30 min at room temperature, it was quenched with saturated NaHCO3 solution. The organic layer was separated, and the aqueous layer was extracted with CH2Cl2 three times. The combined organic layer was dried over MgSO4, and concentrated in vacuo to afford the imidate derivative as a yellow syrup. The imidate derivative was dried over P2O5 under vacuum for 1 d, and it was dissolved in CH2Cl2 (20 mL). 1026) (866 mg, 1.00 mmol) and MS 4 Å (4.0 g) were added to the solution of imidate derivative at room temperature. Next, TMSOTf (20.0 µL, 0.100 mmol) was slowly added to the mixture at −20°C, which was stirred for 45 min at −20°C. The reaction was quenched with saturated NaHCO3 solution at 0°C. The organic layer was separated and washed with saturated NaHCO3 solution and then brine. The organic layer was dried over MgSO4 and concentrated in vacuo. The residue was purified by silica gel chromatography (hexane–AcOEt=2 : 1) to give 15 as colorless syrup (1.47 g, 0.820 mmol, 82%). 15: [α]D21 +14.3 (c=1.0, CHCl3); FT-IR (neat) 3376, 3064, 3029, 2925, 2854, 1741 cm−1; 1H-NMR (500 MHz in CDCl3) δ: 0.87 (12H, m, CH3×4), 1.26 (72H, m, CH2×36), 1.82 (1H, m, β-CH of C3′-O-acyl), 2.30 (2H, m, α-CH2 of N-acyl), 2.35 (2H, m, α-CH2 of C3′-O-acyl), 2.45 (1H, dd, J=15.5, 5.0 Hz, α-CH2 of C3-O-acyl), 2.63 (1H, dd, J=15.5, 8.5 Hz, α-CH2 of C3-O-acyl), 3.70 (10H, m, H-2, H-4, H-5, H-6a, H-2′, H-6′ab, β-CH×2 and OCH2CH=CH2), 4.02 (1H, dd, J=12.5, 5.5 Hz, H-6b), 4.08 (1H, d, J=10.0 Hz, OCH2CH=CH2), 4.27 (1H, dt, J=9.5, 3.5 Hz, H-5′), 4.50 (4H, m, PhCH2×2), 4.68 (5H, m, H-4′, PhCH2 and CH2 of Troc), 4.76 (1H, d, J=4.0 Hz, H-1), 5.09 (9H, m, H-3, H-1′, o-C6H4(CH2O)2P, OCH2CH=CH2 and TrocNH), 5.43 (1H, t, J=10.0 Hz, H-3′), 5.72 (1H, m, OCH2CH=CH2), 6.24 (1H, d, J=9.5 Hz, acylNH), 7.16–7.38 (19H, m, o-C6H4(CH2O)2P and PhCH2×3); 13C-NMR (100 MHz in CDCl3) δ: 14.12, 22.67, 25.05, 25.27, 26.50, 29.35, 29.61, 29.64, 29.67, 31.91, 33.39, 33.58, 33.98, 34.03, 34.38, 38.62, 39.81, 41.73, 51.32, 56.28, 68.27, 68.39, 68.59, 68.65, 68.85, 70.74, 71.06, 71.38, 71.49, 73.62, 74.11, 74.18, 74.54, 74.70, 74.76, 75.73, 76.29, 95.24, 96.55, 100.83, 117.93, 127.56, 127.60, 127.63, 127.66, 127.91, 127.94, 128.32, 128.35, 128.38, 128.42, 128.51, 128.97, 133.35, 134.56, 134.60, 137.79, 138.19, 138.40, 153.98, 171.01, 172.58, 173.41; MS (FAB+) m/z 1805 [M+K]+; HR-MS (FAB+) m/z Calcd for C96H146N2O19Cl3PK: 1805.8960. Found 1805.8953.
Allyl 6-O-{2′-Amino-6′-O-benzyl-2′-deoxy-3′-O-(3-nonyldodecanoyl)-4′-O-(3-oxido-1,5-dihydrobenzo[e][1,3,2]dioxaphosphepin-3-yl)-β-D-glucopyranosyl}-2-deoxy-3-O-[(R)-3-(benzyloxy)tetradecanoyl]-2-[(R)-3-(benzyloxy)tetradecanoylamino]-α-D-glucopyranoside (16)Compound 15 (1.10 g, 0.620 mmol) was dissolved in acetic acid (16 mL). Zinc-copper couple powder (4.40 g, 400% (w/w)) was added to the mixture, which was stirred for 2 h at room temperature. After the suspension was filtrated by Celite®, the filtrate was cooled in ice bath. One molar KOH solution was added dropwise to the mixture at 0°C. The mixture was diluted with AcOEt, and the organic layer was separated and washed with saturated NaHCO3 solution and then brine. The combined organic layer was dried over Na2SO4 and concentrated in vacuo. The residue was purified by silica gel chromatography (hexane–AcOEt–NEt3=1 : 4 : 0.01) to give 16 as colorless syrup (959 mg, 0.601 mmol, 97%). 16: [α]D21 +14.1 (c=1.0, CHCl3); FT-IR (neat) 3370, 3063, 3029, 2925, 2854, 1738 cm−1; 1H-NMR (300 MHz in CDCl3) δ: 0.88 (12H, m, CH3×4), 1.25 (72H, m, CH2×36), 1.94 (1H, m, β-CH of C3′-O-acyl), 2.31 (2H, m, α-CH2 of N-acyl), 2.39 (2H, d, J=6.9 Hz, α-CH2 of C3′-O-acyl), 2.45 (1H, dd, J=15.0, 4.8 Hz, α-CH2 of C3-O-acyl), 2.63 (1H, dd, J=15.0, 7.5 Hz, α-CH2 of C3-O-acyl), 2.90 (1H, dd, J=10.2, 8.1 Hz, H-2′), 3.77 (8H, m, H-2, H-4, H-5, H6′ab, OCH2CH=CH2 and β-CH×2), 4.03 (1H, dd, J=12.6, 5.4 Hz, H-6a), 4.12 (1H, m, OCH2CH=CH2), 4.31 (2H, m, H-6b and H-5′), 4.53 (5H, m, H-4′, PhCH2×2), 4.56 (1H, d, J=12.0 Hz, PhCH2), 4.65 (1H, d, J=12.0 Hz, PhCH2), 4.78 (1H, d, J=3.6 Hz, H-1), 5.08 (9H, m, H-3, H-1′, H-3′, o-C6H4(CH2O)2P and OCH2CH=CH2), 5.72 (1H, m, OCH2CH=CH2), 6.26 (1H, d, J=9.6 Hz, acylNH), 7.15–7.39 (19H, m, o-C6H4(CH2O)2P and PhCH2×3); 13C-NMR (125 MHz in CDCl3) δ: 14.13, 22.68, 25.10, 25.29, 26.40, 26.60, 29.35, 29.62, 29.67, 30.00, 31.91, 33.60, 33.79, 33.99, 34.12, 34.59, 39.00, 39.80, 41.71, 51.26, 56.07, 68.18, 68.23, 68.33, 68.57, 68.64, 68.96, 69.07, 69.38, 70.84, 71.04, 71.40, 73.59, 74.25, 74.29, 74.95, 75.00, 75.76, 76.29, 76.77, 96.57, 104.58, 117.89, 127.59, 127.60, 127.62, 127.86, 127.91, 128.33, 128.36, 128.47, 128.91, 128.92, 133.33, 134.59, 134.63, 137.95, 138.17, 138.39, 171.06, 172.57, 173.64; MS (FAB+) m/z 1616 [M+Na]+; HR-MS (FAB+) m/z Calcd for C93H145N2O17PNa: 1616.0152. Found 1616.0352.
Allyl 6-O-{6′-O-Benzyl-2′-deoxy-3′-O-(3-nonyldodecanoyl)-2′-(3-nonyldodecanoylamino)-4′-O-(3-oxido-1,5-dihydrobenzo[e][1,3,2]dioxaphosphepin-3-yl)-β-D-glucopyranosyl}-2-deoxy-3-O-[(R)-3-(benzyloxy)tetradecanoyl]-2-[(R)-3-(benzyloxy)tetradecanoylamino]-α-D-glucopyranoside (17)To a solution of 16 (924 mg, 0.580 mmol) in CH2Cl2 (62 mL) were added 3-nonyldodecanoic acid (400 mg, 1.20 mmol), MS 4 Å (10.0 g) and EDCI (240 mg, 1.20 mmol) at room temperature. After the reaction mixture was stirred for 11 h at room temperature, it was filtrated by Celite®. The filtrate was diluted with CH2Cl2 and quenched by H2O. The organic layer was separated and washed with brine. The combined organic layer was dried over MgSO4, and concentrated in vacuo. The residue was purified by silica gel chromatography (hexane–AcOEt=2 : 1) to give 17 as colorless syrup (550 mg, 0.290 mmol, 50%). 17: [α]D21 +10.9 (c=1.0, CHCl3); FT-IR (neat) 3514, 3311, 3063, 3031, 2954, 2922, 2852, 1736, 1651 cm−1; 1H-NMR (300 MHz in CDCl3) δ: 0.88 (18H, m, CH3×6), 1.25 (104H, m, CH2×52), 1.81 (2H, m, β-CH of N′-acyl and β-CH of C3′-O-acyl), 2.01 (2H, d, J=7.2 Hz, α-CH2 of N-acyl), 2.31 (4H, m, α-CH2 of N′-acyl and α-CH2 of C3′-O-acyl), 2.43 (1H, dd, J=15.3, 5.4 Hz, α-CH2 of C3-O-acyl), 2.63 (1H, dd, J=15.3, 7.2 Hz, α-CH2 of C3-O-acyl), 3.77 (10H, m, H-2, H-4, H-5, H-6a, H-2′, H-6′ab, OCH2CH=CH2, β-CH of N-acyl and β-CH of C3-O-acyl), 4.01 (2H, m, H-6b and OCH2CH=CH2), 4.28 (1H, dt, J=10.5, 3.3 Hz, H-5′), 4.45 (1H, d, J=11.4 Hz, PhCH2), 4.50 (2H, s, PhCH2), 4.57 (1H, d, J=11.4 Hz, PhCH2), 4.58 (3H, m, H-4′ and PhCH2), 4.76 (1H, d, J=3.6 Hz, H-1), 4.85 (1H, d, J=8.4 Hz, H-1′), 5.06 (7H, m, H-3′, o-C6H4(CH2O)2P and OCH2CH=CH2), 5.39 (1H, dd, J=10.5, 9.3 Hz, H-3), 5.46 (1H, d, J=8.4 Hz, N′H), 5.71 (1H, m, OCH2CH=CH2), 6.21 (1H, d, J=9.3 Hz, NH), 7.15–7.34 (19H, m, o-C6H4(CH2O)2P and PhCH2×3); 13C-NMR (75 MHz in CDCl3) δ: 14.14, 22.71, 25.18, 25.28, 25.62, 26.41, 26.47, 26.55, 26.59, 29.39, 29.43, 29.72, 29.78, 30.07, 30.14, 31.95, 33.26, 33.47, 33.71, 34.09, 38.17, 38.75, 39.78, 41.86, 51.62, 54.59, 67.98, 68.21, 68.43, 68.94, 70.01, 71.14, 71.43, 72.00, 73.61, 73.79, 74.08, 74.72, 75.66, 76.30, 96.52, 100.79, 117.78, 127.53, 127.62, 127.68, 127.82, 128.02, 128.27, 128.33, 128.39, 128.46, 128.53, 128.97, 133.37, 134.64, 134.69, 137.68, 138.39, 138.46, 148.87, 160.52, 171.00, 172.39, 173.15, 173.52; MS (FAB+) m/z 1940 [M+K]+; HR-MS (FAB+) m/z Calcd for C114H185N2O18PK: 1940.2997. Found 1940.3008.
6-O-{6′-O-Benzyl-2′-deoxy-3′-O-(3-nonyldodecanoyl)-2′-(3-nonyldodecanoylamino)-4′-O-(3-oxido-1,5-dihydrobenzo[e][1,3,2]dioxaphosphepin-3-yl)-β-D-glucopyranosyl}-2-deoxy-3-O-[(R)-3-(benzyloxy)tetradecanoyl]-2-[(R)-3-(benzyloxy)tetradecanoylamino]-α-D-glucopyranose (18)To a solution of 17 (500 mg, 0.260 mmol) in THF (43 mL, degassed with Ar) was added [Ir(cod)(MePh2P)2]PF6 (20.0 mg, 0.0260 mmol) at room temperature. After the mixture was stirred for 10 min under bubbling H2 gas at room temperature, it was stirred for another hour. H2O (10 mL) and I2 (66.0 mg, 0.540 mmol) were added to the mixture, which was stirred for 30 min at room temperature. The mixture was diluted with AcOEt and quenched with saturated Na2S2O3 solution. The organic layer was separated and washed with saturated NaHCO3 solution and then brine. The combined organic layer was dried over MgSO4, and concentrated in vacuo. The residue was purified by silica gel chromatography (hexane–AcOEt=2 : 1) to give 18 (421 mg, 0.226 mmol, 87%). 18: [α]D18 −1.9 (c=1.0, CHCl3); FT-IR (neat) 3409, 3327, 3063, 3030, 2954, 2925, 2853, 1742, 1650 cm−1; 1H-NMR (300 MHz in CDCl3) δ: 0.86 (18H, m, CH3×6), 1.23 (104H, m, CH2×52), 1.79 (2H, m, β-CH of N′-acyl and β-CH of C3′-O-acyl), 2.00 (2H, d, J=6.9 Hz, α-CH2 of N-acyl), 2.31 (4H, m, α-CH2 of N′-acyl and α−CH2 of C3′-O-acyl), 2.42 (1H, dd, J=14.7, 4.8 Hz, α-CH2 of C3-O-acyl), 2.59 (1H, dd, J=14.7, 7.8 Hz, α-CH2 of C3-O-acyl), 3.30 (2H, m, β-CH of N-acyl and β−CH of C3-O-acyl), 3.74 (6H, m, H-4, H-5, H-6a, H-2′ and H-6′ab), 3.98 (3H, m, H-2, H-6b and H-5′), 4.52 (7H, m, PhCH2×3 and H-4′), 5.01 (6H, m, H-1, H-1′ and o-C6H4(CH2O)2P), 5.42 (1H, d, J=8.1 Hz, N′H), 5.60 (2H, m, H-3 and H-3′), 6.21 (1H, d, J=9.3 Hz, NH), 7.16–7.37 (19H, m, o-C6H4(CH2O)2P and PhCH2×3); 13C-NMR (100 MHz in CDCl3) δ: 14.13, 22.70, 25.03, 25.10, 25.22, 26.35, 26.42, 26.54, 26.62, 26.70, 29.36, 29.41, 29.64, 29.66, 29.69, 29.72, 29.79, 29.94, 30.04, 30.09, 30.11, 30.19, 31.92, 31.95, 33.29, 33.37, 33.48, 33.73, 33.86, 33.91, 34.07, 34.17, 34.39, 35.10, 38.72, 39.78, 41.90, 51.59, 56.30, 68.28, 68.62, 68.71, 69.70, 70.98, 71.43, 71.56, 71.65, 73.57, 73.77, 74.70, 75.05, 75.11, 75.81, 76.61, 91.56, 99.57, 127.74, 127.81, 127.86, 127.89, 127.98, 128.03, 128.12, 128.35, 128.38, 128.41, 128.46, 128.53, 128.99, 134.66, 137.62, 138.11, 138.44, 171.20, 172.70, 173.13, 174.15; MS (FAB+) m/z 1900 [M+K]+; HR-MS (FAB+) m/z Calcd for C111H181N2O18PK: 1900.2684. Found 1900.2678.
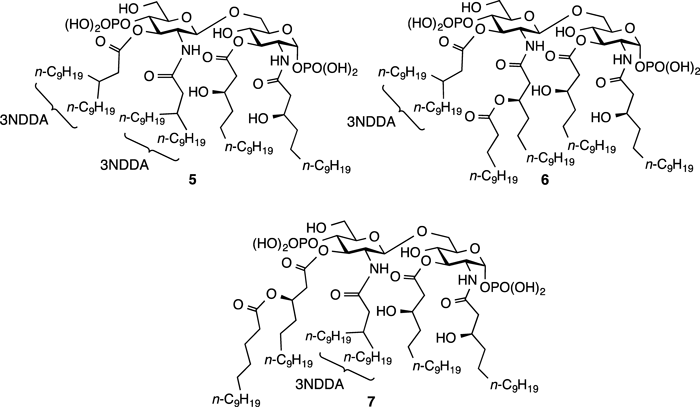
6-O-{2′-Deoxy-3′-O-(3-nonyldodecanoyl)-2′-(3-nonyldodecanoylamino)-β-D-glucopyranosyl}-2-deoxy-3-O-[(R)-3-hydroxytetradecanoyl]-2-[(R)-3-hydroxytetradecanoylamino]-α-D-glucopyranose 4,4′-Diphosphate (5)Compound 18 (370 mg, 0.200 mmol) was dissolved in THF (20 mL), and then 1.6 M solution of n-BuLi (0.16 mL in hexane, 0.260 mmol) was added to the solution at −78°C. The mixture was stirred for 5 min at −78°C, and pyrophosphate (130 mg, 0.240 mmol) was added to the mixture at −78°C. After stirring for 30 min at room temperature, the reaction was quenched with saturated NaHCO3 solution. The organic layer was separated and dried over MgSO4, and it was concentrated to afford the crude product as a yellow syrup. Next, it was dissolved in THF (20 mL), and then Pd-Black (300 mg, 95% (w/w)) was added to the solution at room temperature. The mixture was stirred for 4 h under H2 (10.0 kg/cm2) at room temperature. Triethylamine was added to the mixture at room temperature, and Pd/C was removed by filtration. The filtrate was concentrated in vacuo and the residue was purified by reverse phase silica gel chromatography (MeOH only) to give 5 (213 mg, 0.136 mmol, 68%).
N,O-3NDDA-Dicondenced Analog 5[α]D20 +23.2 (c=1.0, CHCl3); FT-IR (neat) 3360, 3062, 2957, 2933, 2855, 2710, 2500, 1739, 1731, 1660 cm−1; 1H-NMR (300 MHz in C5D5N) δ: 0.87 (18H, m, CH3×6), 1.23 (106H, m, CH2×52, β-CH of N′-acyl and β-CH of C3′-O-acyl), 2.23 (1H, m, α-CH2 of N′-acyl), 2.36 (1H, m, α-CH2 of C3′-O-acyl), 2.69 (6H, m, α-CH2 of N-acyl, α-CH2 of C3-O-acyl, α-CH2 of N′-acyl and α-CH2 of C3′-O-acyl), 3.68 (1H, d, J=9.3 Hz, H-5′), 4.04 (1H, t, J=9.6 Hz, H-4), 4.13 (1H, d, J=12.0 Hz, H-6′a), 4.42 (4H, m, H-6ab, β-CH of N-acyl and β-CH of C3-O-acyl), 4.57 (1H, d, J=12.0 Hz, H-6′b), 4.73 (1H, q, J=9.0 Hz, H-2′), 4.90 (2H, m, H-2 and H-5), 5.01 (1H, q, J=9.9 Hz, H-4′), 5.72 (1H, d, J=8.7 Hz, H-1′), 5.96 (2H, m, H-3 and H-3′), 6.30 (1H, dd, J=7.8, 3.0 Hz, H-1), 8.28 (1H, d, J=9.0 Hz, NH), 9.39 (1H, d, J=9.0 Hz, N′H); 13C-NMR (125 MHz in C5D5N) δ: 8.94, 14.29, 14.32, 22.94, 22.97, 23.00, 26.25, 26.79, 27.14, 27.19, 29.63, 29.70, 29.75, 29.77, 29.87, 29.94, 30.01, 30.05, 30.09, 30.11, 30.15, 30.21, 30.30, 30.40, 30.52, 30.60, 30.70, 32.14, 32.18, 32.23, 33.83, 34.02, 34.10, 34.15, 34.81, 35.63, 38.05, 38.61, 39.90, 41.91, 43.98, 45.56, 45.65, 53.13, 53.69, 54.79, 61.44, 68.24, 68.43, 69.53, 71.97, 74.86, 75.22, 75.35, 77.33, 93.67, 101.76, 172.74, 173.39, 173.46; MS (FAB−) m/z 1568 [M−H]−; HR-MS (FAB−) m/z Calcd for C82H157N2O21P2: 1568.0754. Found 1568.0743.
Allyl 6-O-{6′-O-Benzyl-2′-deoxy-3′-O-(3-nonyldodecanoyl)-2′-[(R)-3-(dodecanoyloxy)tetradecanoylamino]-4′-O-(3-oxido-1,5-dihydrobenzo[e][1,3,2]dioxaphosphepin-3-yl)-β-D-glucopyranosyl}-2-deoxy-3-O-[(R)-3-(benzyloxy)tetradecanoyl]-2-[(R)-3-(benzyloxy)tetradecanoyl Amino]-α-D-glucopyranoside (20)Compound 20 (750 mg, 56%) was prepared from 16 (1.30 g, 0.67 mmol) and (R)-3-(tetradecanoyloxy)tetradecanoic acid (19) (600 mg, 1.30 mmol) according to the procedure described for compound 17. 20: [α]D20 +13.0 (c=1.0, CHCl3); FT-IR (neat) 3514, 3308, 3087, 3065, 3031, 2955, 2925, 2853, 1739, 1653 cm−1; 1H-NMR (500 MHz in CDCl3) δ: 0.88 (18H, m, CH3×6), 1.25–1.58 (112H, m, CH2×56), 1.81 (1H, m, β-CH of C3′-O-acyl), 2.31 (8H, m, α-CH2 of N-acyl, α-CH2 of C3′-O-acyl and N′HCOCH2CHROCOCH2R), 2.42 (1H, m, α-CH2 of C3-O-acyl), 2.63 (1H, dd, J=15.5, 7.5 Hz, α-CH2 of C3-O-acyl), 3.77 (10H, m, H-2, H-4, H-5, H-6a, H-2′, H-6′ab, OCH2CH=CH2, β-CH of N-acyl and β-CH of C3-O-acyl), 4.02 (2H, m, OCH2CH=CH2 and H−6b), 4.28 (1H, dt, J=10.5, 3.5 Hz, H-5′), 4.45 (1H, d, J=11.5 Hz, PhCH2), 4.50 (5H, m, PhCH2×2 and H−4′), 4.54 (1H, d, J=11.5 Hz, PhCH2), 4.77 (1H, d, J=3.5 Hz, H-1), 4.86 (1H, d, J=8.0 Hz, H-1′), 5.11 (8H, m, H-3′, OCH2CH=CH2, o-C6H4(CH2O)2P and β-CH of N′-acyl), 5.39 (1H, dd, J=10.5, 9.0 Hz, H-3), 5.71 (1H, m, OCH2CH=CH2), 6.00 (1H, d, J=8.0 Hz, N′H), 6.22 (1H, d, J=9.5 Hz, NH), 7.16–7.38 (19H, m, o-C6H4(CH2O)2P and PhCH2×3); 13C-NMR (100 MHz in CDCl3) δ: 14.12, 22.69, 24.95, 25.19, 25.26, 25.29, 26.55, 29.25, 29.38, 29.54, 29.58, 29.65, 29.70, 29.78, 30.00, 30.08, 30.11, 31.92, 31.94, 33.41, 33.64, 34.09, 34.32, 34.43, 34.48, 34.57, 38.61, 39.75, 41.88, 42.03, 45.61, 51.62, 51.92, 54.96, 58.72, 67.22, 67.84, 68.26, 68.61, 70.80, 71.17, 71.21, 71.44, 71.90, 73.59, 73.90, 74.14, 74.82, 75.62, 76.29, 96.63, 100.69, 117.77, 127.50, 127.53, 127.62, 127.65, 127.79, 127.95, 128.26, 128.34, 128.38, 128.46, 128.52, 128.96, 129.00, 133.41, 134.63, 134.69, 137.79, 138.45, 170.08, 171.01, 172.35, 173.44, 174.26; MS (FAB+) m/z 2040 [M+K]+; HR-MS (FAB+) m/z Calcd for C119H193N2O20PK: 2040.3521. Found 2040.3530.
6-O-{6′-O-Benzyl-2′-deoxy-3′-O-(3-nonyldodecanoyl)-2′-[(R)-3-(dodecanoyloxy)tetradecanoylamino]-4′-O-(3-oxido-1,5-dihydrobenzo[e][1,3,2]dioxaphosphepin-3-yl)-β-D-glucopyranosyl}-2-deoxy-3-O-[(R)-3-(benzyloxy)tetradecanoyl]-2-[(R)-3-(benzyloxy)tetradecanoyl Amino]-α-D-glucopyranose (21)Compound 21 (430 mg, 87%) was prepared from 20 (500 mg, 0.25 mmol) according to the procedure described for compound 18. 21: [α]D22 +15.4 (c=0.26, CHCl3); FT-IR (neat) 3417, 3331, 3087, 3063, 3030, 2954, 2924, 2853, 1741, 1732, 1652 cm−1; 1H-NMR (500 MHz in CDCl3) δ: 0.88 (18H, m, CH3×6), 1.25–1.58 (112H, m, CH2×56), 1.81 (1H, m, β-CH of C3′-O-acyl), 2.29 (8H, m, α-CH2 of N-acyl, α-CH2 of C3′-O-acyl and N′HCOCH2CHROCOCH2R), 2.43 (1H, m, α-CH2 of C3-O-acyl), 2.61 (1H, dd, J=15.0, 7.5 Hz, α-CH2 of C3-O-acyl), 3.38 (1H, t, J=9.0 Hz, β-CH of N-acyl), 3.55 (1H, q, J=8.5 Hz, β-CH of C3-O-acyl), 3.79 (6H, m, H-4, H-5, H-6a, H-2′ and H-6′ab), 4.01 (2H, m, H-2 and H-6b), 4.19 (1H, dt, J=10.0, 3.5 Hz, H-5′), 4.53 (7H, m, H-4′ and PhCH2×3), 5.07 (7H, m, H-1, H-1′, o-C6H4(CH2O)2P and β-CH of N′-acyl), 5.17 (1H, t, J=8.5 Hz, H-3′), 5.46 (1H, t, J=9.0 Hz, H-3), 6.11 (1H, d, J=8.0 Hz, N′H), 6.29 (1H, d, J=9.5 Hz, NH), 7.14–7.41 (19H, m, o-C6H4(CH2O)2P and PhCH2×3); 13C-NMR (125 MHz in CDCl3) δ: 14.10, 22.68, 24.94, 25.11, 25.21, 25.24, 25.59, 26.56, 29.22, 29.35, 29.49, 29.57, 29.65, 29.68, 29.71, 29.77, 30.01, 30.09, 30.12, 31.92, 33.44, 33.67, 34.13, 34.35, 34.45, 34.49, 38.62, 39.79, 41.89, 42.09, 51.65, 55.57, 67.95, 68.06, 68.28, 68.62, 68.72, 69.56, 70.98, 71.26, 71.52, 71.74, 71.81, 73.59, 74.13, 74.53, 74.98, 75.79, 76.50, 91.60, 99.86, 127.60, 127.66, 127.77, 127.81, 127.85, 127.93, 127.99, 128.33, 128.38, 128.44, 128.49, 128.93, 128.97, 134.67, 134.73, 137.81, 138.16, 138.48, 170.40, 171.18, 172.68, 173.32, 174.58; MS (FAB+) m/z 2000 [M+K]+; HR-MS (FAB+) m/z Calcd for C116H189N2O20PK: 2000.3208. Found 2000.3236.
6-O-{2′-Deoxy-3′-O-(3-nonyldodecanoyl)-2′-[(R)-3-(dodecanoyloxy)tetradecanoylamino]-β-D-glucopyranosyl}-2-deoxy-3-O-[(R)-3-hydroxytetradecanoyl]-2-[(R)-3-hydroxytetradecanoylamino]-α-D-glucopyranose 4,4′-Diphosphate (6)Compound 6 (120 mg, 45%) was prepared from 21 (320 mg, 0.16 mmol) according to the procedure described for compound 5.
O-3NDDA-monocondenced Analog 6[α]D20 +31.0 (c=1.0, CHCl3); FT-IR (neat) 3361, 3064, 2955, 2924, 2853, 2685, 1732, 1660 cm−1; 1H-NMR (300 MHz in C5D5N) δ: 0.87 (18H, m CH3×6), 1.26 (111H, m, CH2×55, β-CH of C3′-O-acyl), 2.22 (1H, m, α-CH2 of C3′-O-acyl), 2.75 (8H, m, α-CH2 of N-acyl, α-CH2 of C3-O-acyl, α-CH2 of C3′-O-acyl and NHCOCH2CHROCOCH2R), 3.21 (1H, dd, J=14,7, 5.1 Hz, N′HCOCH2CHROCOCH2R), 3.71 (1H, d, J=9.9 Hz, H-5′), 3.99 (1H, t, J=9.6 Hz, H-4), 4.16 (1H, d, J=12.0 Hz, H-6′a), 4.45 (5H, m, H-6ab, H-6′b, β-CH of N-acyl and β-CH of C3-O-acyl), 4.71 (1H, q, J=9.9 Hz, H-2′), 4.90 (2H, m, H-2 and H-5), 5.05 (1H, q, J=9.3 Hz, H-4′), 5.78 (1H, d, J=7.8 Hz, H-1′), 5.87 (1H, t, J=5.4 Hz, N′HCOCH2CHROCOCH2R), 5.97 (2H, m, H-3 and H-3′), 6.34 (1H, m, H-1), 8.37 (1H, d, J=7.5 Hz, NH), 9.69 (1H, d, J=8.4 Hz, N′H); 13C-NMR (125 MHz in C5D5N) δ: 8.73, 14.29, 18.67, 22.94, 22.96, 22.99, 23.68, 25.48, 25.82, 26.05, 26.24, 26.77, 27.06, 29.55, 29.63, 29.70, 29.75, 29.82, 29.92, 29.95, 29.99, 30.02, 30.12, 30.20, 30.22, 30.50, 30.60, 30.77, 32.14, 32.18, 32.21, 33.75, 34.07, 34.80, 38.04, 38.45, 39.83, 41.31, 43.92, 45.32, 45.64, 53.45, 54.87, 61.36, 68.21, 68.47, 69.44, 71.52, 72.06, 74.73, 75.23, 77.22, 93.73, 101.52, 170.49, 172.71, 173.35, 173.45; MS (FAB−) m/z 1668 [M−H]+; HR-MS (FAB−) m/z Calcd for C87H165N2O23P2: 1668.1278. Found 1668.1261.
Allyl 6-O-{6′-O-Benzyl-2′-deoxy-3′-O-[(R)-3-(tetradecanoyloxy)tetradecanoyl]-2′-(3-nonyldodecanoylamino)-4′-O-(3-oxido-1,5-dihydrobenzo[e][1,3,2]dioxaphosphepin-3-yl)-β-D-glucopyranosyl}-2-deoxy-3-O-[(R)-3-(benzyloxy)tetradecanoyl]-2-[(R)-3-(benzyloxy)tetradecanoylamino]-α-D-glucopyranoside (23)Compound 23 (1.01 g, 73%) was prepared from 2226) (1.40 g, 0.740 mmol) and 3NDDA (500 mg, 1.5 mmol) according to the procedure described for compound 17. 23: [α]D23 +18.9 (c=1.0, CHCl3); FT-IR (neat) 3517, 3305, 3087, 3063, 3030, 2922, 2852, 1736, 1651 cm−1; 1H-NMR (300 MHz in CDCl3) δ: 0.88 (18H, m, CH3×6), 1.25 (116H, m, CH2×58), 1.82 (1H, m, β-CH of N′-acyl), 2.03 (2H, m, α-CH2 of N-acyl), 2.27 (4H, m, α-CH2 of N′-acyl and OCOCH2CHROCOCH2R), 2.43 (1H, dd, J=15.3, 5.4 Hz, α-CH2 of C3-O-acyl), 2.63 (3H, m, α-CH2 of C3-O-acyl and OCOCH2CHROCOCH2R), 3.74 (10H, m, H-2, H-4, H-5, H-6a, H-2′, H-6′ab, OCH2CH=CH2, β-CH of N-acyl and β-CH of C3-O-acyl), 4.03 (2H, m, H-6b and OCH2CH=CH2), 4.28 (1H, dt, J=10.8, 3.9 Hz, H-5′), 4.46 (1H, d, J=11.4 Hz, PhCH2), 4.53 (1H, d, J=11.4 Hz, PhCH2), 4.55 (5H, m, H-4′ and PhCH2×2), 4.77 (1H, d, J=3.6 Hz, H-1), 5.12 (8H, m, H-1′, H-3′, OCH2CH=CH2, o-C6H4(CH2O)2P and OCOCH2CHROCOCH2R), 5.44 (1H, dd, J=10.2, 9.0 Hz, H-3), 5.72 (1H, m, OCH2CH=CH2), 5.99 (1H, d, J=7.5 Hz, N′H), 6.23 (1H, d, J=9.3 Hz, NH), 7.17–7.38 (19H, m, o-C6H4(CH2O)2P and PhCH2×3); 13C-NMR (75 MHz in CDCl3) δ: 14.13, 22.70, 25.09, 25.19, 25.23, 25.28, 26.39, 26.44, 29.21, 29.37, 29.43, 29.59, 29.68, 29.72, 29.78, 30.04, 30.11, 31.94, 33.24, 33.47, 34.08, 34.58, 34.80, 39.76, 41.61, 41.86, 51.62, 55.51, 68.22, 68.54, 68.85, 70.26, 70.99, 71.17, 71.43, 72.82, 73.61, 73.80, 75.67, 76.31, 96.54, 100.72, 117.77, 127.53, 127.62, 127.65, 127.82, 127.97, 128.27, 128.32, 128.38, 128.49, 128.78, 129.04, 133.38, 134.72, 134.79, 137.74, 138.37, 138.45, 170.35, 171.00, 172.41, 173.60, 173.80; MS (FAB+) m/z 2068 [M+K]+; HR-MS (FAB+) m/z Calcd for C121H197N2O20PK: 2068.3835. Found 2068.3826.
6-O-{6′-O-Benzyl-2′-deoxy-3′-O-[(R)-3-(tetradecanoyloxy)tetradecanoyl]-2′-(3-nonyldodecanoylamino)-4′-O-(3-oxido-1,5-dihydrobenzo[e][1,3,2]dioxaphosphepin-3-yl)-β-D-glucopyranosyl}-2-deoxy-3-O-[(R)-3-(benzyloxy)tetradecanoyl]-2-[(R)-3-(benzyloxy)tetradecanoylamino]-α-D-glucopyranose (24)Compound 24 (490 mg, 82%) was prepared from 23 (600 mg, 0.300 mmol) according to the procedure described for compound 18. 24: [α]D23 −10.5 (c=1.0, CHCl3); FT-IR (neat) 3412, 3320, 3087, 3064, 3031, 2954, 2924, 2852, 1738, 1725, 1649 cm−1; 1H-NMR (500 MHz in CDCl3) δ: 0.88 (18H, m, CH3×6), 1.25 (116H, m, CH2×58), 1.79 (1H, m, β-CH of N′-acyl), 2.02 (2H, t, J=6.0 Hz, α-CH2 of N-acyl), 2.29 (4H, m, α-CH2 of N′-acyl and OCOCH2CHROCOCH2R), 2.44 (1H, dd, J=15.0, 5.0 Hz, α-CH2 of C3-O-acyl), 2.58 (1H, dd, J=13.0, 7.5 Hz, OCOCH2CHROCOCH2R), 2.62 (1H, dd, J=12.0, 7.5 Hz, OCOCH2CHROCOCH2R), 2.66 (1H, dd, J=15.0, 4.0 Hz, α-CH2 of C3-O-acyl), 3.27 (1H, q, J=8.0 Hz, β-CH of N-acyl), 3.37 (1H, t, J=9.5 Hz, β-CH of C3-O-acyl), 3.77 (6H, m, H-4, H-5, H-6a, H-2′ and H-6′ab), 4.02 (2H, m, H-2, H-6b), 4.20 (1H, dt, J=9.5, 3.0 Hz, H-5′), 4.55 (7H, m, H-4′ and PhCH2×3), 5.06 (7H, m, H-1, H-1′, H-3′ and o-C6H4(CH2O)2), 5.27 (1H, m, OCOCH2CHROCOCH2R), 5.59 (1H, t, J=9.0 Hz, H-3), 5.68 (1H, d, J=8.5 Hz, N′H), 6.25 (1H, m, NH), 7.17–7.41 (19H, m, o-C6H4(CH2O)2P and PhCH2×3); 13C-NMR (125 MHz in CDCl3) δ: 14.11, 22.69, 22.71, 25.09, 25.12, 25.23, 26.34, 26.58, 29.23, 29.37, 29.42, 29.56, 29.59, 29.64, 29.66, 29.68, 29.72, 29.78, 30.06, 30.15, 31.93, 31.95, 33.40, 33.52, 34.12, 34.21, 34.62, 34.81, 34.94, 39.80, 40.26, 41.77, 41.96, 51.65, 57.03, 67.97, 68.35, 68.69, 69.74, 70.61, 70.98, 71.59, 71.98, 72.61, 73.64, 73.73, 74.87, 75.15, 75.85, 76.54, 91.61, 99.17, 127.62, 127.67, 127.72, 127.82, 127.89, 127.97, 128.03, 128.10, 128.35, 128.36, 128.41, 128.48, 128.78, 129.01, 134.75, 134.81, 137.73, 138.16, 138.49, 170.12, 171.19, 172.75, 173.72, 174.65; MS (FAB+) m/z 2028 [M+K]+; HR-MS (FAB+) m/z Calcd for C118H193N2O20PK: 2028.3521. Found 2028.3512.
6-O-{2′-Deoxy-3′-O-[(R)-3-(tetradecanoyloxy)tetradecanoyl]-2′-(3-nonyldodecanoylamino)-β-D-glucopyranosyl}-2-deoxy-3-O-[(R)-3-hydroxytetradecanoyl]-2-[(R)-3-hydroxytetradecanoylamino]-α-D-glucopyranose 4,4′-Diphosphate (7)Compound 7 (120 mg, 35%) was prepared from 24 (390 mg, 0.200 mmol) according to the procedure described for compound 5.
N-3NDDA-monocondenced Analog 7[α]D23 +22.3 (c=1.0, CHCl3); FT-IR (neat) 3376, 2954, 2929, 2853, 2699, 1739, 1659, 1652 cm−1; 1H-NMR (300 MHz in C5D5N) δ: 0.86 (18H, m, CH3×6), 1.25 (115H, m, CH2×57, β-CH of N′-acyl), 2.33 (1H, m, α-CH2 of N′-acyl), 2.60 (7H, m, α-CH2 of N-acyl, α-CH2 of C3-O-acyl, α-CH2 of N′-acyl and OCOCH2CHROCOCH2R), 3.04 (1H, m, OCOCH2CHROCOCH2R), 3.30 (1H, dd, J=16.5, 6.6 Hz, OCOCH2CHROCOCH2R), 3.70 (1H, m, H-5), 4.01 (1H, t, J=9.6 Hz, H-4), 4.16 (1H, d, J=12.0 Hz, H-6′a), 4.38 (4H, m, H-6a, H-6′b, β-CH of N-acyl and β-CH of C3-O-acyl), 4.56 (1H, d, J=12.3 Hz, H-6b), 4.77 (1H, dd, J=9.3 Hz, H-2′), 4.91 (2H, m, H-2 and H-5), 5.05 (1H, q, J=9.6 Hz, H-4′), 5.77 (2H, m, H-1′ and OCOCH2CHROCOCH2R), 5.96 (2H, m, H-3 and H-3′), 6.33 (1H, dd, J=7.5, 3.0 Hz, H-1), 8.34 (1H, d, J=9.3 Hz, NH), 9.44 (1H, d, J=8.7 Hz, N′H); 13C-NMR (75 MHz in C5D5N) δ: 8.72 11.57, 14.26, 14.41, 22.78, 22.93, 25.48, 25.63, 25.79, 26.21, 27.12, 27.22, 29.61, 29.74, 29.83, 29.98, 30.09, 30.25, 30.36, 30.50, 30.66, 30.84, 31.99, 32.12, 32.22, 33.83, 33.97, 34.07, 34.39, 34.57, 34.70, 34.89, 35.57, 38.03, 38.34, 38.47, 39.35, 39.83, 41.72, 41.88, 42.06, 43.42, 43.57, 43.90, 45.32, 45.54, 45.69, 51.25, 53.48, 54.50, 54.71, 61.42, 68.12, 68.23, 68.47, 68.57, 68.80, 69.47, 69.82, 70.49, 71.94, 72.97, 74.83, 75.20, 76.12, 76.40, 77.14, 93.12, 93.81, 101.25, 101.64, 170.88, 172.71, 173.28, 173.34, 173.54; MS (FAB−) m/z 1696 [M−H]−; HR-MS (FAB−) m/z Calcd for C89H169N2O23P2: 1696.1591. Found 1696.1578.
6-O-{2′-Deoxy-3′-O-(3-pentyloctanoyl)-2′-(3-pentyloctanoylamino)-β-D-glucopyranosyl}-2-deoxy-3-O-[(R)-3-hydroxytetradecanoyl]-2-[(R)-3-hydroxytetradecanoylamino]-α-D-glucopyranose 4,4′-Diphosphate (25)Compound 25 was synthesized by using 3-pentyloctanoic acid instead of 3NDDA in accordance with the procedure described for compound 5. 25: [α]D19 +24.0 (c=1.0, CHCl3), FT-IR (neat) 3360, 2955, 2931, 2855, 2678, 1738, 1666 cm−1; 1H-NMR (500 MHz in C5D5N) δ: 0.86 (18H, m, CH3×6), 1.25 (74H, m, CH2×36, β-CH of N′-acyl and β-CH of C3′-O-acyl), 2.15 (1H, m, α-CH2 of N′-acyl), 2.28 (1H, m, α-CH2 of C3′-O-acyl), 2.67 (6H, m, α-CH2 of N-acyl, α-CH2 of C3-O-acyl, α-CH2 of N′-acyl and α-CH2 of C3′-O-acyl), 3.64 (1H, d, J=10.0 Hz, H-5′), 4.05 (1H, t, J=9.5 Hz, H-4), 4.10 (1H, d, J=11.5 Hz, H-6′a), 4.40 (4H, m, H-6ab, β-CH of N-acyl and β-CH of C3-O-acyl), 4.54 (1H, d, J=11.5 Hz, H-6′b), 4.65 (1H, q, J=9.0 Hz, H-2′), 4.86 (2H, m, H-2 and H-5), 4.93 (1H, dd, J=10.0, 9.5 Hz, H-4′), 5.66 (1H, d, J=8.5 Hz, H-1′), 5.86 (1H, t, J=9.5 Hz, H-3′), 5.95 (1H, t, J=10.0 Hz, H-3), 6.25 (1H, dd, J=8.0, 3.0 Hz, H-1), 8.26 (1H, d, J=8.5 Hz, NH), 9.30 (1H, d, J=9.0 Hz, N′H); 13C-NMR (75 MHz in C5D5N) δ: 8.78, 14.24, 14.31, 14.34, 22.90, 22.96, 23.00, 23.06, 23.07, 26.20, 26.23, 26.28, 26.66, 26.68, 26.71, 29.58, 29.90, 29.95, 29.98, 30.06, 30.08, 32.09, 32.51, 32.53, 32.57, 32.66, 33.60, 33.82, 33.91, 33.95, 34.64, 35.46, 38.00, 38.39, 39.70, 41.75, 43.92, 45.21, 45.66, 53.40, 53.45, 54.72, 61.30, 68.13, 68.40, 69.25, 71.94, 74.82, 74.86, 75.20, 77.14, 93.84, 101.80, 172.67, 173.24, 173.30, 173.33; MS (FAB−) m/z 1343 [M−H]−; HR-MS (FAB−) m/z Calcd for C66H125N2O21P2: 1343.8250. Found 1343.8238.
6-O-{2′-Deoxy-3′-O-(3-heptyldecanoyl)-2′-(3-heptyldecanoylamino)-β-D-glucopyranosyl}-2-deoxy-3-O-[(R)-3-hydroxytetradecanoyl]-2-[(R)-3-hydroxytetradecanoylamino]-α-D-glucopyranose 4,4′-Diphosphate (26)Compound 26 was synthesized by using 3-heptyldecanoic acid instead of 3NDDA in accordance with the procedure described for compound 5. 26: [α]D19 +21.3 (c=1.0, CHCl3), FT-IR (neat) 3370, 2955, 2925, 2854, 2728, 1737, 1658 cm−1; 1H-NMR (500 MHz in C5D5N) δ: 0.85 (18H, m, CH3×6), 1.28 (90H, m, CH2×44, β-CH of N′-acyl and C3′-O-acyl), 2.19 (1H, m, α-CH2 of N′-acyl), 2.32 (1H, m, α-CH2 of C3′-O-acyl), 2.70 (6H, m, α-CH2 of N-acyl, α-CH2 of C3-O-acyl, α-CH2 of N′-acyl and α-CH2 of C3′-O-acyl), 3.68 (1H, d, J=9.0 Hz, H-5′), 4.03 (1H, t, J=9.5 Hz, H-4), 4.14 (1H, d, J=13.0 Hz, H-6′a), 4.38 (4H, m, H-6ab, β-CH of N-acyl and β-CH of C3-O-acyl), 4.54 (1H, d, J=11.5 Hz, H-6′b), 4.74 (1H, m, H-2′), 4.90 (2H, dd, J=9.0, 8.0 Hz, H-2 and H-5), 5.04 (1H, dd, J=10.0, 9.5 Hz, H-4′), 5.72 (1H, d, J=8.5 Hz, H-1′), 5.99 (2H, m, H-3 and H-3′), 6.34 (1H, dd, J=7.5, 3.0 Hz, H-1), 8.44 (1H, d, J=8.5 Hz, NH), 9.38 (1H, d, J=9.0 Hz, N′H); 13C-NMR (125 MHz in C5D5N) δ: 8.61, 14.25, 14.28, 14.31, 22.90, 22.96, 22.98, 26.22, 26.70, 26.86, 27.03, 27.09, 27.22, 29.60, 29.65, 29.68, 29.71, 29.74, 29.79, 29.82, 29.91, 29.97, 30.20, 30.34, 30.37, 30.42, 30.54, 32.10, 32.15, 32.19, 32.25, 33.76, 33.98, 34.05, 34.72, 35.53, 38.06, 38.16, 38.32, 39.77, 41.91, 43.92, 45.09, 45.66, 53.31, 53.37, 54.69, 61.32, 68.14, 68.21, 68.49, 68.53, 69.33, 72.37, 72.41, 74.63, 75.14, 77.01, 94.11, 94.14, 101.78, 172.64, 173.29, 173.39, 173.44; MS (FAB−) m/z 1455 [M−H]; HR-MS (FAB−) m/z Calcd for C74H141N2O21P2: 1455.9502. Found 1455.9531.
6-O-{2′-Deoxy-3′-O-(3-undecyltetradecanoyl)-2′-(3-undecyltetradecanoylamino)-β-D-glucopyranosyl}-2-deoxy-3-O-[(R)-3-hydroxytetradecanoyl]-2-[(R)-3-hydroxytetradecanoylamino]-α-D-glucopyranose 4,4′-Diphosphate (27)Compound 27 was synthesized by using 3-undecyltetradecanoic acid instead of 3NDDA in accordance with the procedure described for compound 5. 27: [α]D20 +17.4 (c=1.0, CHCl3); FT-IR (neat) 3367, 2955, 2932, 2855, 2737, 2622, 2605, 2530, 2498, 1738, 1665 cm−1; 1H-NMR (500 MHz in C5D5N) δ: 0.88 (18H, m, CH3×6), 1.25 (122H, m, CH2×60, β-CH of N′-acyl and β-CH of C3′-O-acyl), 2.24 (1H, m, α-CH2 of N′-acyl), 2.36 (1H, m, α-CH2 of C3′-O-acyl), 2.64 (6H, m, α-CH2 of N-acyl, α-CH2 of C3-O-acyl, α-CH2 of N′-acyl and α-CH2 of C3′-O-acyl), 3.67 (1H, d, J=9.0 Hz, H-5′), 4.06 (1H, dd, J=10.0, 9.0 Hz, H-4), 4.13 (1H, d, J=12.0 Hz, H-6′a), 4.41 (4H, m, H-6ab, β-CH of N-acyl and β-CH of C3-O-acyl), 4.57 (1H, d, J=12.0 Hz, H-6′b), 4.74 (1H, dd, J=9.5, 8.5 Hz, H-2′), 4.90 (2H, m, H-2 and H-5), 5.01 (1H, q, J=10.0 Hz, H-4′), 5.49 (1H, d, J=8.5 Hz, H-1′), 5.97 (2H, m, H-3 and H-3′), 6.31 (1H, dd, J=8.0, 3.0 Hz, H-1), 8.31 (1H, d, J=9.0 Hz, NH), 9.41 (1H, d, J=8.5 Hz, N′H); 13C-NMR (125 MHz in C5D5N) δ: 8.61, 14.28, 14.30, 22.94, 22.97, 26.24, 26.26, 26.78, 27.13, 29.63, 29.67, 29.70, 29.95, 30.01, 30.04, 30.06, 30.08, 30.13, 30.17, 30.24, 30.32, 30.51, 30.55, 30.60, 30.71, 32.13, 32.15, 32.19, 33.80, 34.01, 39.86, 41.94, 43.97, 45.32, 45.64, 53.50, 54.74, 61.36, 68.24, 68.48, 69.40, 72.19, 74.69, 75.17, 75.27, 77.17, 79.83, 93.92, 101.85, 172.71, 173.34, 173.47; MS (FAB+) m/z 1704 [M+Na]+; HR-MS (FAB+) m/z Calcd for C87H174N2O21P2Na: 1704.1982. Found 1704.1995.
6-O-{2′-Deoxy-3′-O-(3-tridecylhexadecanoyl)-2′-(3-tridecylhexadecanoylamino)-β-D-glucopyranosyl}-2-deoxy-3-O-[(R)-3-hydroxytetradecanoyl]-2-[(R)-3-hydroxytetradecanoylamino]-α-D-glucopyranose 4,4′-Diphosphate (28)Compound 28 was synthesized by using 3-tridecylhexadecanoic acid instead of 3NDDA in accordance with the procedure described for compound 5. 28: [α]D20 +16.9 (c=1.0, CHCl3); FT-IR (neat) 3373, 2924, 2853, 2679, 2498, 1739, 1659 cm−1; 1H-NMR (500 MHz in C5D5N) δ: 0.88 (18H, m, CH3×6), 1.25 (138H, m, CH2×68, β-CH of N′-acyl and β-CH of C3′-O-acyl), 2.24 (1H, m, α-CH2 of N′-acyl), 2.37 (1H, m, α-CH2 of C3′-O-acyl), 2.69 (6H, m, α-CH2 of N-acyl, α-CH2 of C3-O-acyl, α-CH2 of N′-acyl and α-CH2 of C3′-O-acyl), 3.68 (1H, d, J=9.0 Hz, H-5′), 4.05 (1H, dd, J=10.0, 9.5 Hz, H-4), 4.14 (1H, d, J=12.5 Hz, H-6′a), 4.42 (4H, m, H-6ab, β-CH of N-acyl and β-CH C3-O-acyl), 4.57 (1H, d, J=12.5 Hz, H-6′b), 4.75 (1H, q, J=9.0 Hz, H-2′), 4.91 (2H, m, H-2 and H-5), 5.03 (1H, q, J=10.0 Hz, H-4′), 5.74 (1H, d, J=8.0 Hz, H-1′), 5.98 (2H, m, H-3 and H-3′), 6.33 (1H, dd, J=8.0, 3.0 Hz, H-1), 8.33 (1H, d, J=8.5 Hz, NH), 9.42 (1H, d, J=9.0 Hz, N′H); 13C-NMR (125 MHz in C5D5N) δ: 8.74, 14.42, 23.07, 23.09, 26.09, 26.38, 26.90, 27.25, 29.76, 29.80, 30.08, 30.10, 30.14, 30.17, 30.22, 30.23, 30.28, 30.33, 30.36, 30.39, 30.45, 30.64, 30.67, 30.74, 30.84, 32.27, 32.29, 33.92, 34.14, 34.24, 34.89, 35.71, 38.21, 38.57, 39.99, 42.07, 44.07, 45.42, 45.80, 53.60, 54.87, 61.46, 68.36, 68.60, 69.51, 72.29, 74.83, 75.22, 75.37, 77.26, 79.46, 79.72, 79.93, 79.98, 94.06, 101.99, 172.84, 173.47, 173.61; MS (FAB+) m/z 1816 [M]+; HR-MS (FAB+) m/z Calcd for C98H190N2O21P2: 1816.3234. Found 1816.3251.
Docking Simulation AnalysisThe docking study of 5 was carried out using Molecular Operating Environment software (MOE 2014.09, Chemical Computing Group, Montreal, Canada). The X-ray crystallographic structures of MD2 (PDB ID: 2E59) and TLR4/MD2 complex (PDB ID: 3FXI) were used. The minimizations of MD2 and TLR4/MD2 complex were performed using MMFF94s force-field. The active sites of MD2 and TLR4/MD2 complex were identified using MOE a Site Finder. The docking simulations with 5 and these proteins (Fig. 6 and graphic abstract) were performed using ASE Dock program (Ryoka Systems, Tokyo, Japan).