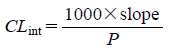Experimental
In Vitro TGR5 AssayhTGR5/CRE/HEK293 or mTGR5/CRE/HEK293 stable cell line was obtained by transfection of HEK293 cells with human or mouse TGR5 expression plasmid (hTGR5-pcDNA3.1 or mTGR5-pcDNA3.1) and CRE-driven luciferase reporter plasmid (pGL4.29; Promega, Madison, WI, U.S.A.), and employed to assess the activity of test compounds by reporter gene assay. Briefly, cells were seeded into 96-well plates and incubated overnight in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) in 5% CO2 at 37°C. Then, cells were incubated with fresh medium containing different concentrations of test compounds or 20 µM INT-777 as a positive control for 5.5 h. Luciferase activity in cell lysate was determined using the Steady-Glo Luciferase Assay System (Promega) according to the manufacturer’s instructions. Compounds with EC50 values on human TGR5 (hTGR5) less than 100 nM were selected for the mouse TGR5 (mTGR5) activity test.
In Vitro Microsomal Assay for Metabolic StabilityThe test compound was dissolved in dimethyl sulfoxide (DMSO) and diluted to the desired concentration with DMSO and 0.1% aqueous bovine serum albumin (BSA). Human or mouse liver microsomes were incubated in a 96-well plate containing 0.1 M Tris buffer (pH 7.4), 0.33 mg/mL microsomal protein, 0.1 µM test compound, 5.0 mM MgCl2, 0.005% BSA, and 1.0 mM reduced nicotinamide adenine dinucleotide phosphate (NADPH). Incubations were conducted at 37°C for 60 min. An aliquot was removed at each time point (0, 7, 17, 30, 60 min) and the enzymatic reaction was stopped by protein precipitation in methanol. The loss of the test compound was determined by LC-MS/MS. The intrinsic clearance (CLint, mL/min/g microsomal protein) was calculated from the following equations.
C0: concentration at time 0;
t: time (min);
k: slope (min
−1).
P: microsomal protein concentration (mg/mL).
Synthetic Materials and MethodsUnless otherwise noted, all materials were purchased from commercial suppliers and used without further purification. All reaction yields were not optimized. Analytical TLC (0.15–0.2 mm) and preparative TLC (0.4–0.5 mm) were performed on HSGF254 (Yantai Jiangyou Company, Yantai, Shandong, China), compounds were visualized by UV light (254 nm). Column chromatography was carried out on silica gel (200−300 mesh) or with pre-packed silica cartridges (4–40 g) from Bonna-Agela Technologies Inc. (Tianjin, China) and eluted with system of the Teledyne Isco CombiFlash Rf. Microwave reactions were performed in a Biatage Inititor. Melting point of target compounds was measured by SGWX-4 melting point apparatus (Shanghai Precision and Scientific Instrument Corporation, Shanghai, China). 1H- and 13C-NMR spectra were recorded on a Bruker AC300 or a Bruker AC400 or a Bruker AC500 NMR spectrometer, using tetramethylsilane (TMS) as an internal reference. 1H-NMR date are reported as multiplicity (s singlet, d doublet, t triplet, q quarter, dd doublet of doublets, dt doublet of triplets, br s broad singlet, m multiplet), number of protons, and coupling constant in hertz (Hz). Spectra were carried in CDCl3, DMSO-d6, and CD3OD. IR spectra were recorded on IS5 FT-IR (Thermo). Low-resolution mass spectra were determined on an Agilent LC-MS systems that consisted of an Agilent 1260 infinity LC coupled to an Agilent 6120 Quadrupole mass spectrometer (electrospray ionization (ESI)+) using an Agilent (Symmetry® C18 Colum, 3.5 µm, 2.1×50 mm) with aqueous CH3CN (0.4 mL/min, 0–5 min, 30–90%; 5–9 min, 90–90%) containing 0.1% formic acid and monitored at λ 240 nm. High-resolution (HR) mass spectra were recorded on a Q-TOF Ultima Globe mass spectrameter (Micromass, Manchester, U.K.).
5-(2,5-Dimethylphenoxy)-1,3-dimethyl-1H-pyrazole-4-carbaldehyde (11c)Compound 10 (500 mg, 3.15 mmol) was dissolved in N,N-dimethylformamide (DMF) and treated with excess 2,5-dimethylphenol (1.15 g, 9.43 mmol) and potassium hydroxide (KOH) (270 mg, 4,82 mmol), then the reaction mixture was stirred at 120°C for 2 h. After consumption of 10 as monitored by TLC, the mixture was cooled to room temperature, then poured into water and extracted three times with EtOAc. The combined organic phases were washed with brine (3×), dried over MgSO4, filtered, and concentrated to dryness. The residue was purified by flash column chromatography to give 11c (661 mg, 86%). 1H-NMR (300 MHz, CDCl3) δ: 9.33 (1H, s), 7.13 (1H, d, J=5.7 Hz), 6.89 (1H, d, J=5.7 Hz,), 6.53 (1H, s), 3.64 (3H, s), 2.45 (3H, s), 2.32 (3H, s), 2.24 (3H, s).
5-(2,5-Dimethylphenoxy)-1,3-dimethyl-1H-pyrazole-4-carboxylic Acid (12c)To a solution of 11c (0.62 g, 2.54 mmol) in mixed of tertiary butanol (9 mL), tetrahydrofuran (THF) (9 mL) and H2O (3 mL), was added NaH2PO4 (1.83 g, 15.2 mmol), NaClO2 (1.38 g, 15.2 mmol) and 2-methylbut-2-ene (4 mL), and then allowed to stir at room temperature (r.t.) for 3 h. After completion of the reaction, the solvent was removed by rotary evaporation, and the crude was suspended in water. The precipitate was collected via filtration to yield 12c (0.60 g, 92%). 1H-NMR (300 MHz, DMSO) δ: 12.01 (1H, br), 7.14 (1H, d, J=7.8 Hz), 6.80 (1H, d, J=7.8 Hz), 6.25 (1H, s), 3.52 (3H, s), 2.32 (3H, s), 2.28 (3H, s), 2.16 (3H, s).
(4-Cyclopropyl-3,4-dihydroquinoxalin-1(2H)-yl)(5-(2,5-dimethylphenoxy)-1,3-dimethyl-1H-pyrazol-4-yl)methanone (13c)To a solution of 12c (0.5 g, 1.92 mmol) in dry dichloromethane (DCM) was added oxalyl chloride (491 µL, 5.76 mmol) and three drops DMF, and held at reflux for 2 h. Then the reaction mixture was cooled to room temperature and concentrated. The residue was re-dissolved in dry DCM, 1-cyclopropyl-1,2,3,4-tetrahydroquinoxaline (341 mg, 1.92 mmol) and Et3N (797 µL, 5.76 mmol) were added, and the reaction was stirred at r.t. overnight. After completion, the reaction was diluted with water and extracted with DCM (3×). The combined organic phases were collected, washed with brine, dried over MgSO4, concentrated and purified by flash column chromatography to afford the title compound 13c (624 mg) as an off-white solid in 78% yield. mp 124°C. 1H-NMR (300 MHz, CDCl3) δ: 7.07 (1H, dd, J=0.9 Hz, 6.3 Hz), 6.99–6.94 (2H, m), 6.75 (2H, d, J=5.7 Hz), 6.49 (1H, dt, J=0.9, 6.0 Hz), 6.40 (1H, s), 3.73 (2H, m), 3.49 (3H, s), 2.91 (2H, m), 2.30 (1H, m), 2.26 (3H, s), 2.20 (3H, s), 2.01 (3H, s), 0.74 (2H, m), 0.45 (2H, m). 13C-NMR (126 MHz, CDCl3) δ: 162.07, 154.12, 148.23, 146.18, 139.16, 137.04, 131.01, 125.41, 125.27, 123.82, 123.07, 122.55, 116.28, 113.34, 112.97, 103.61, 48.36, 40.66, 34.15, 31.27, 21.12, 15.55, 13.62, 7.89. IR (KBr) cm−1: 1626.27, 1504.57, 1419.43, 1404.60, 1361.74, 738.51. HPLC-MS: 5.66 min, 417.3 (M+H+). HR-MS (ESI): m/z (M+H+) Calcd for C25H29O2N4: 417.2285. Found: 417.2275.
(4-Cyclopropyl-3,4-dihydroquinoxalin-1(2H)-yl)(1,3-dimethyl-5-phenoxy-1H-pyrazol-4-yl)methanone (13a)The title compound 13a was prepared from 10 and phenol according to a procedure similar to that described for the preparation of the title compound 13c. mp 145°C. 1H-NMR (300 MHz, CDCl3) δ: 7.24 (2H, t, J=6.0 Hz), 7.13 (1H, d, J=5.7 Hz), 7.05 (1H, d, J=5.7 Hz), 7.00 (1H, d, J=6.3 Hz), 6.73 (2H, d, J=6.0 Hz), 6.70 (1H, d, J=6.3 Hz), 6.48 (1H, t, J=5.7 Hz), 3.72 (2H, m), 3.53 (3H, s), 2.85 (2H, m), 2.30 (1H, m), 2.28 (3H, s), 0.78 (2H, m), 0.51 (2H, m). 13C-NMR (126 MHz, CDCl3) δ: 161.89, 156.36, 148.53, 145.96, 139.18, 129.77, 125.41, 125.24, 123.45, 122.64, 116.24, 115.04, 112.76, 103.29, 48.25, 40.50, 34.20, 31.25, 13.61, 7.99. IR (KBr) cm−1: 1621.84, 1502.90, 1489.58, 1411.74, 1400.72, 1320.10, 756.37. HPLC-MS: 4.93 min, 389.3 (M+H+). HR-MS (ESI): m/z (M+H+) Calcd for C23H25O2N4: 389.1972. Found: 389.1965.
(4-Cyclopropyl-3,4-dihydroquinoxalin-1(2H)-yl)(5-(2,5-dichlorophenoxy)-1,3-dimethyl-1H-pyrazol-4-yl)methanone (13b)The title compound 13b was obtained as a white solid from 10 and 2,5-dichlorophenol according to the general procedure. mp 139°C. 1H-NMR (300 MHz, CDCl3) δ: 7.15 (1H, d, J=7.8 Hz), 7.00 (1H, dt, J=1.2, 7.8 Hz), 6.72 (1H, d, J=7.8 Hz), 6.62 (1H, d, J=8.7 Hz), 6.49 (1H, t, J=7.8 Hz), 6.30 (1H, d, J=2.4 Hz), 6.18 (1H, dd, J=2.4, 8.7 Hz), 3.76 (2H, m), 3.52 (3H, s), 3.02 (2H, m), 2.35 (1H, m), 2.22 (3H, s), 0.80 (2H, m), 0.55 (2H, m). 13C-NMR (126 MHz, CDCl3) δ: 165.04, 161.57, 156.72, 151.78, 148.00, 145.43, 139.52, 133.40, 131.21, 125.81, 124.49, 122.84, 120.85, 116.53, 116.00, 113.19, 48.91, 41.19, 34.49, 31.26, 13.44, 7.98. HPLC-MS: 5.76 min, 457.2 (M+H+). HR-MS (ESI): m/z (M+H+) Calcd for C23H23O2N4Cl2: 457.1193. Found: 457.1186.
(4-Cyclopropyl-3,4-dihydroquinoxalin-1(2H)-yl)(5-(3-methoxyphenoxy)-1,3-dimethyl-1H-pyrazol-4-yl)methanone (13d)The title compound 13d was prepared from 10 and 3-methoxyphenol following the general procedure. mp 109°C. 1H-NMR (300 MHz, CDCl3) δ: 7.15 (2H, m), 6.99 (1H, t, J=8.1 Hz), 6.71 (1H, d, J=7.5 Hz), 6.61 (1H, m), 6.48 (1H, m), 6.31 (2H, m), 3.77 (2H, m), 3.74 (3H, s), 3.52 (3H, s), 2.90 (2H, m), 2.30 (1H, m), 2.25 (3H, s), 0.78 (2H, m), 0.50 (2H, m). 13C-NMR (126 MHz, CDCl3) δ: 160.84, 157.34, 151.14, 148.44, 139.24, 130.30, 130.20, 125.40, 122.65, 116.24, 112.80, 109.02, 108.96, 107.13, 106.96, 101.78, 55.44, 48.26, 40.98, 34.22, 31.20, 22.70, 13.60, 7.99. HPLC-MS: 4.99 min, 419.3 (M+H+). HR-MS (ESI): m/z (M+H+) Calcd for C24H27O3N4: 419.2078. Found: 419.2072.
Ethyl-5-chloro-1-methyl-1H-pyrazole-4-carboxylate (15)To a solution of tBuONO (2.95 mL, 2.46 mmol) in acetonitrile (ACN) was added CuCl (1.76 g, 1.77 mmol), and then added 14 (2.5 g, 1.48 mmol) to the mixture in portions. The reaction was stirred at r.t. for 2 h, and then heated at 60°C for 1 h. After completion as TLC monitored, the reaction mixture was cooled to r.t. and diluted with 2 N aq. HCl, then extracted with DCM (3×). The combined phases were washed with brine, dried over MgSO4, filtered, concentrated and purified by flash column chromatography to give 15 (2.08 g) in 75% yield. 1H-NMR (300 MHz, CDCl3) δ: 7.90 (1H, s), 4.30 (2H, q, J=5.4 Hz), 3.86 (3H, s), 1.34 (3H, t, J=5.4 Hz).
Ethyl-1-methyl-5-phenoxy-1H-pyrazole-4-carboxylate (16a)In a microwave reaction vessel was combined 15 (300 mg, 1.59 mmol), phenol (300 mg, 3.19 mmol) and Cs2CO3 (1.52 g, 4.66 mmol) in DMF. The vessel was sealed and the resulting mixture was heated at 120°C by microwave for 30 min. Then the reaction mixture was cooled to r.t., quenched with water and extracted with EtOAc three times. The organic layer was collected, washed with brine, dried over MgSO4 and filtered. After the solvent was removed, the crude was purified by flash column chromatography to yield 16a (280 mg, 72%). 1H-NMR (300 MHz, CDCl3) δ: 7.92 (1H, s), 7.3 (2H, dt, J=2.1, 7.2 Hz), 7.10 (1H, dt, J=1.2, 7.2 Hz), 6.89 (2H, m), 4.07 (2H, q, J=7.2 Hz), 3.71 (3H, s), 1.03 (3H, t, J=7.2 Hz).
1-Methyl-5-phenoxy-1H-pyrazole-4-carboxylic Acid (17a)16a (270 mg, 1.10 mmol) was dissolved in a solution of 1,4-dioxane–H2O=2 : 1, and NaOH (88 mg, 2.20 mmol) was added. The mixture was stirred at r.t. until consumption of 16a as monitored by TLC, and then the solvent was removed. The residue was re-dissolved in water, acidified with 2 N aqueous HCl to pH 3 and then the product precipitated out. The precipitate was collected by filtration to afford 17a (227 mg, 95%) and was carried forward without further purification. 1H-NMR (300 MHz, DMSO) δ: 12.25 (1H, s), 7.85 (1H, s), 7.36 (2H, dd, J=1.5, 7.5 Hz), 7.12 (1H, dt, J=0.9 Hz, 7.5 Hz), 6.92 (2H, m), 3.61 (3H, s).
(4-Cyclopropyl-3,4-dihydroquinoxalin-1(2H)-yl)(1-methyl-5-phenoxy-1H-pyrazol-4-yl)methanone (18a)The title compound 18a was prepared as an off-white solid from 17a and 1-cyclopropyl-1,2,3,4-tetrahydroquinoxaline according to a procedure similar to the preparation of 13c from 12c. mp 104°C. 1H-NMR (300 MHz, CDCl3) δ: 7.54 (1H, s), 7.26 (2H, m), 7.15 (1H, d, J=8.1 Hz), 7.08 (1H, d, J=7.5 Hz), 7.02 (1H, d, J=8.4 Hz), 6.79 (1H, d, J=7.8 Hz), 6.75 (2H, d, J=8.1 Hz), 6.52 (1H, d, J=7.5 Hz), 3.74 (2H, t, J=5.4 Hz), 3.60 (3H, s), 2.94 (2H, t, J=5.4 Hz), 2.31 (1H, m), 0.77 (2H, m), 0.52 (2H, m). 13C-NMR (126 MHz, CDCl3) δ: 160.92, 156.47, 146.70, 140.20, 139.36, 129.78, 125.67, 125.26, 123.50, 123.21, 116.29, 115.03, 112.86, 105.00, 48.34, 40.60, 34.61, 31.25, 7.97. HPLC-MS: 4.84 min, 375.3 (M+H+). HR-MS (ESI): m/z (M+H+) Calcd for C22H23O2N4: 375.1816. Found: 375.1810.
(4-Cyclopropyl-3,4-dihydroquinoxalin-1(2H)-yl)(5-(2,5-dimethylphenoxy)-1-methyl-1H-pyrazol-4-yl)methanone (18b)The title compound 18b was obtained as a white solid from 15 according to the general procedure that preparation of title compound 18a. mp 131°C. 1H-NMR (300 MHz, CDCl3) δ: 7.52 (1H, s), 7.10 (1H, d, J=8.1 Hz), 6.99 (2H, m), 6.81 (1H, d, J=8.1 Hz), 6.76 (1H, d, J=7.5 Hz), 6.51 (1H, d, J=7.5 Hz), 6.37 (1H, s), 3.76 (2H, t, J=5.4 Hz), 3.57 (3H, s), 3.00 (2H, t, J=5.4 Hz), 2.26 (1H, m), 2.20 (3H, s), 2.06 (3H, s), 0.74 (2H, m), 0.47 (2H, m). 13C-NMR (126 MHz, CDCl3) δ: 161.14, 154.32, 146.95, 140.07, 139.32, 136.95, 131.10, 125.57, 125.45, 123.90, 123.25, 123.17, 116.32, 113.14, 113.02, 105.15, 48.47, 40.72, 34.53, 31.27, 21.13, 15.53, 7.88. HPLC-MS: 5.57 min, 403.3 (M+H+). HR-MS (ESI): m/z (M+H+) Calcd for C24H27O2N4: 403.2129. Found: 403.2122.
(4-Cyclopropyl-3,4-dihydroquinoxalin-1(2H)-yl)(5-(3-methoxyphenoxy)-1-methyl-1H-pyrazol-4-yl)methanone (18c)The title compound 18c was obtained from 15 according to the general procedure that preparation of title compound 18a. mp 58°C. 1H-NMR (300 MHz, CDCl3) δ: 7.53 (1H, s), 7.14 (2H, m), 7.03 (1H, dt, J=0.9, 6.0 Hz), 6.80 (1H, d, J=6.0 Hz), 6.61 (1H, m), 6.51 (1H, dt, J=1.2, 5.7, 6.0 Hz), 6.32 (2H, m), 3.76 (2H, t, J=4.2 Hz), 3.75 (3H, s), 3.60 (3H, s), 2.97 (2H, t, J=4.2 Hz), 2.31 (1H, m), 0.78 (2H, m), 0.53 (2H, m). 13C-NMR (126 MHz, CDCl3) δ: 160.94, 160.85, 157.45, 146.55, 140.16, 139.44, 130.20, 125.69, 125.25, 123.24, 116.30, 112.91, 109.08, 106.88, 105.09, 101.70, 55.41, 48.35, 40.64, 34.62, 31.21, 7.96. IR (KBr) cm−1: 2925.24, 1637.36, 1607.05, 1501.83, 1489.73, 1430.37, 1130.12, 743.44. HPLC-MS: 4.90 min, 405.3 (M+H+). HR-MS (ESI): m/z (M+H+) Calcd for C23H25O3N4: 405.1921. Found: 405.1913.
Ethyl-5-hydroxy-1-(4-methoxybenzyl)-1H-1,2,3-triazole-4-carboxylate (19)Compound 19 was prepared as the literature described.26) 1H-NMR (300 MHz, DMSO) δ: 7.20 (2H, d, J=6.3 Hz), 6.91 (2H, d, J=6.3 Hz), 5.27 (2H, s), 4.24 (2H, q, J=5.4 Hz), 3.72 (1H, s), 1.27 (3H, t, J=5.4 Hz).
Ethyl-5-chloro-1-(4-methoxybenzyl)-1H-1,2,3-triazole-4-carboxylate (20)Compound 20 was synthesized from 19 with the method described in literature.27) 1H-NMR (300 MHz, CDCl3) δ: 7.26 (2H, d, J=8.7 Hz), 6.87 (2H, d, J=8.7 Hz), 5.50 (2H, s), 4.42 (2H, q, J=4.2 Hz), 3.79 (1H, s), 1.40 (3H, t, J=4.2 Hz).
Ethyl-1-(4-methoxybenzyl)-5-(3-methoxyphenoxy)-1H-1,2,3-triazole-4-carboxylate (21b)3-Methoxyphenol (334 µL, 3.04 mmol) was dissolved in dry DMF, then cooled to 0°C and added NaH (60%, 90 mg, 2.25 mmol). The reaction mixture was warmed to r.t. and stirred for 0.5 h. The residue was treated with 20 (600 mg, 2.03 mmol) and held at 85°C. After completion as monitored by TLC, the reaction was cooled to r.t. and slowly quenched with water. The resulting mixture was extracted twice with EtOAc, dried over MgSO4 and concentrated. The crude was purified by flash column chromatography to give 21b (576 mg) in 74% yield. 1H-NMR (300 MHz, CDCl3) δ: 7.20 (2H, d, J=8.7 Hz), 7.14 (1H, t, J=8.7 Hz), 6.78 (2H, d, J=8.7 Hz), 6.64 (1H, dd, J=2.1, 8.7 Hz), 6.32 (2H, m), 5.33 (2H, s), 4.19 (2H, q, J=4.2 Hz), 3.75 (3H, s), 3.71 (3H, s), 1.12 (3H, t, J=4.2 Hz).
1-(4-Methoxybenzyl)-5-(3-methoxyphenoxy)-1H-1,2,3-triazole-4-carboxylic Acid (22b)Compound 22b (502 mg) was prepared from 21b (570 mg, 1.49 mmol) according to the procedure described that preparation of 17a from 16a in excellent yield (95%). 1H-NMR (300 MHz, DMSO) δ: 12.99 (1H, br), 7.19 (1H, t, J=8.4 Hz), 7.14 (2H, t, J=8.7 Hz), 6.83 (2H, d, J=8.7 Hz), 6.69 (1H, ddd, J=0.6, 2.4, 8.7 Hz), 6.42 (1H, t, J=2.4 Hz), 6.36 (1H, ddd, J=0.6, 2.4, 8.7 Hz) 5.35 (2H, s), 3.69 (3H, s), 3.68 (3H, s).
(4-Cyclopropyl-3,4-dihydroquinoxalin-1(2H)-yl)(1-(4-methoxybenzyl)-5-(3-methoxyphenoxy)-1H-1,2,3-triazol-4-yl)methanone (23b)The title compound 23b was obtained as a white solid from 22b following a similar procedure that preparation of 13c from 12c. mp 111°C. 1H-NMR (300 MHz, CDCl3) δ: 7.26–6.97 (6H, m), 6.76 (2H, d, J=7.8 Hz), 6.61 (1H, d, J=7.8 Hz), 6.40 (1H, m), 6.19 (2H, m), 5.23 (2H, s), 3.88 (2H, m), 3.76 (3H, s), 3.69 (3H, s), 2.91 (2H, m), 2.28 (1H, m), 0.76 (2H, m), 0.51 (2H, m). 13C-NMR (126 MHz, CDCl3) δ: 160.78, 159.70, 159.01, 156.67, 139.41, 130.20, 129.66, 129.52, 125.99, 125.90, 124.10, 116.22, 114.12, 112.94, 109.91, 107.24, 101.81, 55.43, 55.31, 50.48, 48.10, 41.44, 31.20, 7.98. HPLC-MS: 5.72 min, 512.3 (M+H+). HR-MS (ESI): m/z (M+H+) Calcd for C29H30O4N5: 512.2292. Found: 512.2285.
(4-Cyclopropyl-3,4-dihydroquinoxalin-1(2H)-yl)(5-(2,5-dichlorophenoxy)-1-(4-methoxybenzyl)-1H-1,2,3-triazol-4-yl)methanone (23a)The title compound 23a was obtained as a white solid with a similar method that preparation of the title compound 23b from 20. mp 119°C. 1H-NMR (300 MHz, CDCl3) δ: 7.26 (2H, m), 7.11 (3H, m), 7.01 (1H, dt, J=1.2, 2.0 Hz), 6.95 (1H, dd, J=2.1, 6.3 Hz), 6.72 (2H, d, J=6.3 Hz), 6.54 (1H, m), 6.36 (1H, m), 5.31 (2H, s), 4.06 (2H, m), 3.75 (3H, s), 3.28 (2H, m), 2.34 (1H, m), 0.77 (2H, m), 0.54 (2H, m). 13C-NMR (126 MHz, CDCl3) δ: 159.84, 158.52, 151.23, 139.65, 133.15, 131.12, 130.27, 129.72, 126.15, 125.20, 124.98, 124.30, 123.64, 121.14, 116.47, 114.20, 113.31, 55.29, 51.14, 48.86, 31.34, 7.97. IR (KBr) cm−1: 1642.80, 1515.46, 1497.38, 1473.21, 1254.21. HPLC-MS: 6.37 min, 550.2 (M+H+). HR-MS (ESI): m/z (M+H+) Calcd for C28H26O3N5Cl2: 550.1407. Found: 550.1403. HR-MS (ESI): m/z (M+H+) Calcd for C28H26O3N5Cl2: 550.1407. Found: 550.1403.
(5-(2,5-Dichlorophenoxy)-1-(4-methoxybenzyl)-1H-1,2,3-triazol-4-yl)(4-methyl-3,4-dihydro-quinoxalin-1(2H)-yl)methanone (24a)In addition to replaced1-cyclopropyl-1,2,3,4-tetrahydroquinoxaline with 1-methyl-1,2,3,4-tetrahydroquinoxaline, the title compound 24a was obtained as a white solid with a similar method that preparation of the title compound 23b from 20. mp 116°C. 1H-NMR (300 MHz, CDCl3) δ: 7.26 (2H, m), 7.12 (2H, m), 7.02–6.94 (2H, m), 6.73 (2H, d, J=8.4 Hz), 6.58 (1H, d, J=8.1 Hz), 6.45 (1H, m), 6.37 (1H, m), 5.30 (2H, s), 4.05 (2H, m), 3.75 (3H, s), 3.19 (2H, m), 2.83 (3H, s). 13C-NMR (126 MHz, CDCl3) δ: 159.83, 158.52, 151.17, 139.43, 133.16, 131.07, 130.09, 129.73, 126.62, 125.24, 124.96, 123.90, 121.17, 116.23, 115.72, 114.20, 111.52, 55.29, 51.14, 50.83, 37.93. IR (KBr) cm−1: 1644.47, 1514.06, 1474.96, 1248.25, 748.35. HPLC-MS: 5.88 min, 524.2 (M+H+). HR-MS (ESI): m/z (M+H+) Calcd for C26H24O3N5: 524.1251. Found: 524.1251.
(1-(4-Methoxybenzyl)-5-(3-methoxyphenoxy)-1H-1,2,3-triazol-4-yl)(4-methyl-3,4-dihydro-quinoxalin-1(2H)-yl)methanone (24b)In addition to replaced 1-cyclopropyl-1,2,3,4-tetrahydroquinoxaline with 1-methyl-1,2,3,4-tetrahydroquinoxaline, the title compound 24b was obtained as a white solid with a similar method that preparation of the title compound 23b from 20. mp 122°C. 1H-NMR (300 MHz, CDCl3) δ: 7.12–7.07 (4H, m), 7.00 (1H, dt, J=1.2, 6.0 Hz), 6.76 (2H, d, J=6.3 Hz), 6.62 (1H, dd, J=1.2, 6.3 Hz), 6.57 (1H, d, J=6.3 Hz), 6.33 (1H, m), 6.19 (2H, m), 5.23 (2H, s), 3.88 (2H, m), 3.76 (3H, s), 3.70 (3H, s), 2.77 (5H, m). 13C-NMR (126 MHz, CDCl3) δ: 160.79, 159.69, 158.95, 156.61, 139.22, 130.10, 129.64, 129.13, 126.42, 126.08, 123.60, 115.33, 114.12, 110.86, 110.01, 107.17, 101.71, 55.44, 55.31, 50.43, 37.77. HPLC-MS: 5.21 min, 486.3 (M+H+). HR-MS (ESI): m/z (M+H+) Calcd for C27H28O4N5: 486.2136. Found: 486.2123.
(5-(3-Methoxyphenoxy)-1H-1,2,3-triazol-4-yl)(4-methyl-3,4-dihydroquinoxalin-1(2H)-yl)methanone (25b)24b (70 mg, 144 µmol) was dissolved in trifluoroacetic acid (TFA) (5 mL), then heated to 65°C and stirred for 2 h. Upon completion of reaction, the reaction mixture was cooled to r.t. and concentrated. The residue was then re-dissolved in DCM and washed with water. Then the solvent was removed under reduced pressure, and the crude reaction mixture was purified by preparative TLC to yield the title compound 25b (35 mg, 66%) as a light yellow solid. mp 157°C. 1H-NMR (300 MHz, CDCl3) δ: 7.18 (1H, d, J=8.4 Hz), 7.02 (1H, d, J=7.8 Hz), 6.61 (3H, m), 6.36 (3H, m), 3.99 (2H, m), 3.73 (3H, s), 3.21 (2H, m), 2.87 (3H, s). HPLC-MS: 3.98 min, 366.2 (M+H+). HR-MS (ESI): m/z (M+H+) Calcd for C19H20O3N5: 366.1561. Found: 366.1556.
(5-(2,5-Dichlorophenoxy)-1H-1,2,3-triazol-4-yl)(4-methyl-3,4-dihydroquinoxalin-1(2H)-yl)methanone (25a)The title compound 25a was prepared as a light yellow solid from 24a according to the similar procedure that preparation of the title compound 25b from 24b. mp 149°C. 1H-NMR (300 MHz, CDCl3) δ: 7.23 (1H, m), 6.97 (1H, m), 6.88 (1H, m), 6.55–6.39 (4H, m), 4.07 (2H, d, J=5.4 Hz), 3.48 (2H, d, J=5.4 Hz), 2.87 (3H, s). 13C-NMR (126 MHz, CDCl3) δ: 150.77, 139.67, 132.79, 130.65, 127.13, 124.97, 123.73, 123.02, 122.27, 118.83, 115.33, 111.24, 50.73, 40.13, 37.90. IR (KBr) cm−1: 1629.60, 1512.67, 1474.02, 1401.87, 1329.41, 1229.21, 733.17. HPLC-MS: 4.99 min, 404.1 (M+H+). HR-MS (ESI): m/z (M+H+) Calcd for C18H16O2N5Cl2: 404.0676. Found: 404.0664.
Ethyl-4-hydroxy-2-methylthiazole-5-carboxylate (26)Compound 26 was synthesized with the method described in the patent.28) 1H-NMR (300 MHz, CDCl3) δ: 9.88 (1H, br), 4.34 (2H, q, J=7.2 Hz), 2.65 (1H, s), 1.35 (3H, t, J=7.2 Hz).
Ethyl-4-(5-chloro-2-nitrophenoxy)-2-methylthiazole-5-carboxylate (27a)To a solution of 26 in ethanol was added dropwise with fresh sodium ethoxide and then allowed to stir at 45°C for 0.5 h. The reaction mixture was cooled to r.t. and filtered to give sodium salt of 26 (2 g, 9.57 mmol). The residue was treated with 4-chloro-2-fluoro-1-nitrobenzene (1.7 g, 9.71 mmol) in NMP and heated by microwave at 125°C for 2 h. Then the reaction mixture was cooled to r.t., poured into water, extracted with EtOAc three times and combined the organic phases. The organic layer was washed with brine twice, dried over MgSO4, filtered and concentrated. The crude was purified by flash column chromatography to give compound 27a (2.21 g) in yield of 65%. 1H-NMR (300 MHz, CDCl3) δ: 8.02 (1H, d, J=8.7 Hz), 7.29 (1H, dd, J=2.1, 8.7 Hz), 7.27 (2H, d, J=2.1 Hz), 4.31 (2H, q, J=7.2 Hz), 2.57 (3H, s), 1.32 (3H, t, J=7.2 Hz).
Ethyl-4-(2-amino-5-chlorophenoxy)-2-methylthiazole-5-carboxylate (28a)To a solution of 27a (1.37 g, 4.0 mmol) in THF–H2O (2 : 1) was added iron powder (2.24 g, 40 mmol) and NH4Cl (2.14g, 40 mmol), and then stirred at 65°C overnight. After completion of the reaction as monitored by TLC, the mixture was cooled to r.t., filtered by siliceousearth and the filter cake was washed with DCM. The filtrate was extracted with DCM and the organic layer was combined. The residue was evaporated under reduced pressure to give crude product 28a and was used directly in the next step without further purification. HPLC-MS: 313.13 (M+H+).
Ethyl-4-(2,5-dichlorophenoxy)-2-methylthiazole-5-carboxylate (29a)To a solution of 28a (312 mg, 1.0 mmol) in ACN was added CuCl2 (202 mg, 1.5 mmol), and slowly added tBuONO (238 µL, 2.0 mmol). The reaction mixture was allowed to stir at r.t. for 24 h, then heated to 60°C and stirring was continued for 2 h. Upon completing, the reaction was quenched with 2 N aqueous HCl and diluted with water. The mixture was extracted with DCM (3×) and the organic phases were combined. The combined organic layer was washed twice with brine, dried over MgSO4 and concentrated. The residue was purified by flash column chromatography to give the product 29a (249 mg, 75%). 1H-NMR (300 MHz, CDCl3) δ: 7.36 (1H, d, J=6.9 Hz), 7.11 (1H, dd, J=1.8, 6.9 Hz), 7.10 (1H, d, J=1.8 Hz), 4.31 (2H, q, J=5.4 Hz), 2.61 (3H, s), 1.32 (3H, t, J=5.4 Hz).
Ethyl-4-(2-bromo-5-chlorophenoxy)-2-methylthiazole-5-carboxylate (29c)In addition to use CuBr2 instead of CuCl2, 29c was synthesized from 28a as following a similar procedure that preparation 29a from 28a. 1H-NMR (300 MHz, CDCl3) δ: 7.52 (1H, d, J=6.0 Hz), 7.04 (2H, m), 4.31 (2H, q, J=5.4 Hz), 2.62 (3H, s), 1.31 (3H, t, J=5.4 Hz).
Ethyl-4-(3-chlorophenoxy)-2-methylthiazole-5-carboxylate (29e)A solution of 28a (312 mg, 1.0 mmol) in ACN, was treated with tBuONO and held at reflux for 8 h. After the reaction was completed, the reaction mixture was cooled to r.t. and quenched with 2 N aqueous HCl, then diluted with water. The crude mixture was extracted with EtOAc, and the combined organic layer was washed twice with brine, dried over MgSO4, filtered and concentrated under reduced pressure. The residue was purified by flash column chromatography to yield 29e (194 mg) in 65%. 1H-NMR (300 MHz, CDCl3) δ: 7.27 (1H, t, J=6.0 Hz), 7.12 (1H, dd, J=1.5, 6.0 Hz), 7.09 (1H, d, J=1.5 Hz), 7.00 (1H, dd, J=1.5, 6.0 Hz), 4.29 (2H, q, J=5.4 Hz), 2.63 (3H, s), 1.30 (3H, t, J=5.1 Hz).
4-(2,5-Dichlorophenoxy)-2-methylthiazole-5-carboxylic AcidThe intermediate was prepared with the same method described that preparation of 17a from 16a. 1H-NMR (300 MHz, DMSO) δ: 13.21 (1H, br), 7.60 (1H, d, J=6.6 Hz), 7.38 (1H, d, J=1.5 Hz), 7.31 (1H, d, J=1.5, 6.6 Hz), 2.55 (3H, s).
4-(2-Bromo-5-chlorophenoxy)-2-methylthiazole-5-carboxylic AcidThe intermediate was prepared with the same method described that preparation of 17a from 16a. 1H-NMR (300 MHz, DMSO) δ: 13.19 (1H, br), 7.60 (1H, d, J=6.6 Hz), 7.38 (1H, d, J=1.5 Hz), 7.31 (1H, d, J=1.5, 6.6 Hz), 2.55 (3H, s).
4-(3-Chlorophenoxy)-2-methylthiazole-5-carboxylic AcidThe intermediate was prepared with the same method described that preparation of 17a from 16a. 1H-NMR (300 MHz, DMSO) δ: 13.18 (1H, br), 7.39 (1H, t, J=6.0 Hz), 7.22 (1H, dd, J=1.5, 6.0 Hz), 7.17 (1H, d, J=1.5 Hz), 7.03 (1H, dd, J=1.5, 6.0 Hz), 2.58 (3H, s).
(4-Cyclopropyl-3,4-dihydroquinoxalin-1(2H)-yl)(4-(2,5-dichlorophenoxy)-2-methylthiazol-5-yl)methanone (30c)The title compound 30c was prepared as a light yellow solid according to the general procedure that preparation of 13c from 12c. mp 160°C. 1H-NMR (400 MHz, CDCl3) δ: 7.21 (1H, d, J=8.6 Hz), 6.95–6.89 (2H, m), 6.85 (1H, dd, J=8.3, 1.5 Hz), 6.79 (1H, d, J=7.9 Hz), 6.52 (1H, ddd, J=8.4, 7.2, 1.5 Hz), 6.13 (1H, s), 3.94 (2H, t, J=5.5 Hz), 3.49 (2H, t, J=5.5 Hz), 2.61 (3H, s), 2.30 (1H, tt, J=6.8, 3.7 Hz), 0.74–0.68 (2H, m), 0.45–0.39 (2H, m). 13C-NMR (101 MHz, CDCl3) δ: 166.75, 159.52, 154.26, 151.09, 139.83, 132.58, 130.40, 126.51, 125.52, 124.65, 122.95, 122.47, 119.93, 116.00, 113.22, 112.84, 48.98, 41.30, 31.22, 20.24, 8.04. IR (KBr) cm−1: 1646.23, 1536.75, 1506.15, 1472.16, 1405.36, 1331.26, 748.47. HPLC-MS: 6.49 min, 460.1 (M+H+). HR-MS (ESI): m/z (M+H+) Calcd for C22H19Cl2N3O2S: 460.0648. Found: 460.0661.
(4-(2,5-Dichlorophenoxy)-2-methylthiazol-5-yl)(4-methyl-3,4-dihydroquinoxalin-1(2H)-yl)methanone (30d)In addition to replaced 1-cyclopropyl-1,2,3,4-tetrahydro-quinoxaline with 1-methyl-1,2,3,4-tetrahydro-quinoxaline, 30d was prepared as a light yellow solid according to the general procedure that preparation of 13c from 12c. mp 126°C. 1H-NMR (300 MHz, CDCl3) δ: 7.19 (1H, d, J=8.7 Hz), 6.90 (1H, dd, J=2.4, 8.4 Hz), 6.84 (1H, dt, J=1.2, 7.8 Hz), 6.74 (1H, d, J=7.2 Hz), 6.44 (1H, dt, J=1.2, 7.2, 7.8 Hz), 6.24 (1H, d, J=7.8 Hz), 6.17 (1H, s), 3.99 (2H, t, J=5.4 Hz), 3.49 (2H, t, J=5.4 Hz), 2.78 (3H, s), 2.63 (3H, s). 13C-NMR (126 MHz, CDCl3) δ: 166.64, 159.32, 153.62, 150.84, 139.48, 132.43, 130.15, 126.81, 124.77, 124.07, 122.92, 121.72, 119.05, 115.08, 113.50, 111.03, 50.78, 41.03, 37.73, 20.13. IR (KBr) cm−1: 1640.10, 1625.80, 1512.29, 1473.01, 1407.68, 1333.55, 742.15. HPLC-MS: 6.01 min, 434.1 (M+H+). HR-MS (ESI): m/z (M+H+) Calcd for C20H18O2N3Cl2S: 434.0491. Found: 434.0487.
(4-(2-Chloro-5-methylphenoxy)-2-methylthiazol-5-yl)(4-cyclopropyl-3,4-dihydroquinoxalin-1(2H)-yl)methanone (30a)The title compound 30a was obtained as a light yellow solid according to the general procedure that preparation of 13c from 12c. mp 136°C. 1H-NMR (300 MHz, CDCl3) δ: 7.31 (1H, dd, J=1.2, 7.2 Hz), 7.03–6.97 (2H, m), 6.92–6.88 (3H, m), 6.51 (1H, m), 6.33 (1H, m), 3.96 (2H, t, J=5.4 Hz), 3.46 (2H, t, J=5.4 Hz), 2.59 (3H, s), 2.32 (1H, m), 0.71 (2H, m), 0.42 (2H, m). 13C-NMR (126 MHz, CDCl3) δ: 166.36, 159.89, 155.34, 150.44, 139.76, 137.49, 129.41, 125.87, 125.69, 125.46, 122.96, 120.98, 120.37, 115.79, 113.15, 111.52, 48.85, 41.70, 31.06, 20.86, 20.11, 7.87. IR (KBr) cm−1: 1628.74, 1506.62, 1415.80, 1332.32, 1067.33, 757.53. HPLC-MS: 6.27 min, 440.2 (M+H+). HR-MS (ESI): m/z (M+H+) Calcd for C23H23O2N3ClS: 440.1194. Found: 440.1189.
(4-(2-Chloro-5-methylphenoxy)-2-methylthiazol-5-yl)(4-methyl-3,4-dihydroquinoxalin-1(2H)-yl)methanone (30b)In addition to replaced 1-cyclopropyl-1,2,3,4-tetrahydro-quinoxaline with 1-methyl-1,2,3,4-tetrahydro-quinoxaline, the title compound 30b was obtained as a light yellow solid according to the general procedure that preparation of 13c from 12c. mp 137°C. 1H-NMR (300 MHz, CDCl3) δ: 7.15 (1H, d, J=8.1 Hz), 6.87–6.82 (2H, m), 6.75 (1H, dd, J=1.2, 8.1 Hz), 6.45 (1H, m), 6.32 (1H, d, J=7.8 Hz), 6.10 (1H, s), 4.00 (2H, t, J=5.4 Hz), 3.46 (2H, t, J=5.4 Hz), 2.79 (3H, s), 2.60 (3H, s), 2.17 (3H, s). 13C-NMR (126 MHz, CDCl3) δ: 166.38, 159.83, 154.93, 150.41, 139.61, 137.46, 129.36, 126.30, 125.10, 123.09, 120.50, 119.75, 118.32, 115.11, 112.21, 111.30, 50.84, 41.58, 37.87, 20.93, 20.12. HPLC-MS: 5.76 min, 414.2 (M+H+). HR-MS (ESI): m/z (M+H+) Calcd for C21H21O2N3ClS: 414.1038. Found: 414.1031.
(4-(2-Bromo-5-chlorophenoxy)-2-methylthiazol-5-yl)(4-cyclopropyl-3,4-dihydroquinoxalin-1(2H)-yl)methanone (30e)The title compound 30e was obtained as a light yellow solid according to the general procedure that preparation of 13c from 12c. mp 144°C. 1H-NMR (300 MHz, CDCl3) δ: 7.37 (1H, d, J=7.2 Hz), 6.94–6.76 (4H, m), 6.52 (1H, dt, J=1.5, 7.5 Hz), 6.04 (1H, s), 3.93 (2H, t, J=5.4 Hz), 3.51 (2H, t, J=5.4 Hz), 2.61 (3H, s), 2.31 (1H, m), 0.71 (2H, m), 0.42 (2H, m). 13C-NMR (126 MHz, CDCl3) δ: 166.64, 159.37, 154.06, 152.08, 139.66, 133.28, 133.19, 126.42, 125.32, 124.90, 122.83, 119.61, 115.78, 113.06, 112.90, 110.79, 48.89, 41.08, 31.14, 20.13, 7.94. IR (KBr) cm−1: 1645.76, 1534.69, 1505.39, 1466.46, 1403.98, 1331.27, 747.58. HPLC-MS: 6.55 min, 504.1 (M+H+). HR-MS (ESI): m/z (M+H+) Calcd for C22H20O2N3BrClS: 504.0143. Found: 504.0130.
(4-(2-Bromo-5-chlorophenoxy)-2-methylthiazol-5-yl)(4-methyl-3,4-dihydroquinoxalin-1(2H)-yl)methanone (30f)In addition to replaced 1-cyclopropyl-1,2,3,4-tetrahydro-quinoxaline with 1-methyl-1,2,3,4-tetrahydro-quinoxaline, the title compound 30e was obtained as a light yellow solid according to the general procedure that preparation of 13c from 12c. mp 134°C. 1H-NMR (300 MHz, CDCl3) δ: 7.35 (1H, d, J=8.4 Hz), 6.86–6.82 (2H, m), 6.73 (1H, d, J=8.1 Hz), 6.44 (1H, d, J=7.8 Hz), 6.21 (1H, d, J=8.4 Hz), 6.10 (1H, s), 3.99 (2H, t, J=5.4 Hz), 3.51 (2H, t, J=5.4 Hz), 2.78 (3H, s), 2.63 (3H, s). 13C-NMR (126 MHz, CDCl3) δ: 166.66, 159.30, 153.57, 151.95, 146.03, 139.46, 133.25, 133.09, 126.86, 124.74, 124.46, 122.93, 118.91, 115.02, 113.71, 111.00, 50.80, 40.98, 37.79, 20.14. HPLC-MS: 6.09 min, 478.1 (M+H+). HR-MS (ESI): m/z (M+H+) Calcd for C20H18O2N3BrClS: 477.9986. Found: 477.9980.
(4-(2-Bromo-5-methylphenoxy)-2-methylthiazol-5-yl)(4-cyclopropyl-3,4-dihydroquinoxalin-1(2H)-yl)methanone (30g)The title compound 30g was obtained as a light yellow solid according to the general procedure that preparation of 13c from 12c. mp 133°C. 1H-NMR (300 MHz, CDCl3) δ: 7.32 (1H, d, J=6.0 Hz), 6.88 (3H, m), 6.71 (1H, dd, J=0.9, 6.0 Hz), 6.53 (1H, m), 5.95 (1H, d, J=0.9 Hz), 3.95 (2H, t, J=4.2 Hz), 3.50 (2H, t, J=4.2 Hz), 2.59 (3H, s), 2.31 (1H, m), 2.15 (3H, s), 0.69 (2H, m), 0.41 (2H, m). 13C-NMR (126 MHz, CDCl3) δ: 166.37, 159.86, 155.25, 151.56, 139.74, 138.26, 132.38, 125.89, 125.66, 122.97, 120.24, 118.57, 115.68, 113.14, 111.76, 109.53, 48.89, 41.56, 31.08, 20.88, 20.13, 7.90. IR (KBr) cm−1: 1628.27, 1526.51, 1505.97, 1480.77, 1413.50, 1331.67, 1308.91, 756.78. HPLC-MS: 6.33 min, 484.2 (M+H+). HR-MS (ESI): m/z (M+H+) Calcd for C23H23O2N3BrS: 484.0689. Found: 484.0683.
(4-(2-Bromo-5-methylphenoxy)-2-methylthiazol-5-yl)(4-methyl-3,4-dihydroquinoxalin-1(2H)-yl)methanone (30h)In addition to replaced 1-cyclopropyl-1,2,3,4-tetrahydro-quinoxaline with 1-methyl-1,2,3,4-tetrahydro-quinoxaline, the title compound 30h was obtained as a light yellow solid according to the general procedure that preparation of 13c from 12c. mp 129°C. 1H-NMR (300 MHz, CDCl3) δ: 7.32 (1H, d, J=6.0 Hz), 6.88 (3H, m), 6.71 (1H, dd, J=0.9, 6.0 Hz), 6.53 (1H, m), 6.10 (1H, s), 3.99 (2H, t, J=5.4 Hz), 3.49 (2H, t, J=5.4 Hz), 2.79 (3H, s), 2.60 (3H, s), 2.13 (3H, s). 13C-NMR (126 MHz, CDCl3) δ: 166.52, 159.81, 154.82, 151.51, 139.61, 138.21, 132.33, 128.75, 126.32, 125.53, 124.93, 122.99, 119.62, 117.76, 115.02, 111.28, 50.85, 41.16, 37.90, 20.96, 20.13. HPLC-MS: 6.16 min, 458.1 (M+H+). HR-MS (ESI): m/z (M+H+) Calcd for C21H21O2N3BrS: 458.0532. Found: 458.0531.
(4-Cyclopropyl-3,4-dihydroquinoxalin-1(2H)-yl)(2-methyl-4-(m-tolyloxy)thiazol-5-yl)methanone (30i)The title compound 30i was obtained ass a light yellow solid according to the general procedure that preparation of 13c from 12c. mp 116°C. 1H-NMR (300 MHz, CDCl3) δ: 7.10 (1H, t, J=7.8 Hz), 7.01–6.91 (3H, m), 6.81 (1H, d, J=7.8 Hz), 6.55–6.50 (2H, m), 6.47 (1H, s), 3.92 (2H, t, J=5.4 Hz), 3.38 (2H, t, J=5.4 Hz), 2.60 (3H, s), 2.30 (1H, m), 0.74 (2H, m), 0.42 (2H, m). 13C-NMR (126 MHz, CDCl3) δ: 166.49, 159.61, 155.78, 154.88, 139.93, 134.37, 129.81, 126.23, 125.85, 123.81, 122.70, 118.58, 116.32, 116.21, 113.21, 112.50, 48.95, 41.51, 31.15, 20.09, 7.92. IR (KBr) cm−1: 1622.93, 1591.20, 1506.27, 1406.76, 1332.96, 1214.85, 1086.47, 746.28. HPLC-MS: 6.18 min, 426.2 (M+H+). HR-MS (ESI): m/z (M+H+) Calcd for C22H21O2N3ClS: 426.1038. Found: 426.1033.
(4-Cyclopropyl-3,4-dihydroquinoxalin-1(2H)-yl)(2-methyl-4-(m-tolyloxy)thiazol-5-yl)methanone (30j)The title compound 30j was obtained as a light yellow solid according to the general procedure that preparation of 13c from 12c. mp 110°C. 1H-NMR (300 MHz, CDCl3) δ: 7.09–6.94 (3H, m), 6.88 (1H, d, J=7.8 Hz), 6.83 (1H, d, J=7.5 Hz), 6.57-6.51 (1H, m), 6.42 (1H, m), 6.34 (1H, s), 3.92 (2H, t, J=5.7 Hz), 3.36 (2H, t, J=5.7 Hz), 2.58 (3H, s), 2.30 (1H, m), 2.24 (3H, s), 0.71 (2H, m), 0.41 (2H, m). 13C-NMR (126 MHz, CDCl3) δ: 166.28, 160.07, 155.99, 155.41, 139.87, 139.19, 128.82, 125.98, 125.91, 124.49, 122.83, 118.68, 116.25, 115.13, 113.19, 111.65, 48.86, 41.83, 31.08, 21.28, 20.10, 7.89. IR (KBr) cm−1: 1623.00, 1506.34, 1405.22, 1334.80, 1306.90, 1248.01, 1146.39, 746.32. HPLC-MS: 5.98 min, 406.2 (M+H+). HR-MS (ESI): m/z (M+H+) Calcd for C23H24O2N3S: 406.1584. Found: 406.1578.
(4-Cyclopropyl-3,4-dihydroquinoxalin-1(2H)-yl)(4-(2,4-dichlorophenoxy)-2-methylthiazol-5-yl)methanone (30k)The title compound 30k was synthesized as a light yellow solid in a manner similar to the title compound 30c. mp 87°C. 1H-NMR (400 MHz, CDCl3) δ: 7.31 (1H, d, J=2.5 Hz), 6.98 (1H, dd, J=8.8, 2.6 Hz), 6.95–6.88 (2H, m), 6.85 (1H, d, J=8.0 Hz), 6.50 (1H, ddd, J=8.2, 6.5, 2.2 Hz), 6.22 (1H, d, J=8.8 Hz), 3.95 (2H, t, J=5.5 Hz), 3.47 (2H, t, J=5.5 Hz), 2.57 (3H, s), 2.33 (1H, tt, J=6.8, 3.7 Hz), 0.77–0.70 (2H, m), 0.46–0.39 (2H, m). 13C-NMR (126 MHz, CDCl3) δ: 166.48, 159.60, 154.91, 149.57, 139.83, 129.58, 129.30, 127.36, 126.12, 125.72, 125.23, 122.77, 121.00, 115.99, 113.18, 111.48, 48.86, 41.56, 31.10, 20.05, 7.97. HPLC-MS: 6.62 min, 460.1 (M+H+). HR-MS (ESI): m/z (M+H+) Calcd for C22H20O2N3Cl2S: 460.0648. Found: 460.0641.
(4-(4-Chlorophenoxy)-2-methylthiazol-5-yl)(4-cyclopropyl-3,4-dihydroquinoxalin-1(2H)-yl)methanone (30l)The title compound 30l was synthesized as a light yellow solid in a manner similar to the title compound 30i. mp 88°C. 1H-NMR (400 MHz, CDCl3) δ: 7.16–7.11 (2H, m), 7.03–6.94 (2H, m), 6.83 (1H, d, J=7.9 Hz), 6.52 (3H, ddd, J=12.6, 7.8, 1.8 Hz), 3.93 (2H, t, J=5.6 Hz), 3.38 (2H, t, J=5.6 Hz), 2.58 (3H, s), 2.31 (1H, tt, J=6.7, 3.7 Hz), 0.77–0.71 (2H, m), 0.44–0.38 (2H, m). 13C-NMR (126 MHz, CDCl3) δ: 166.43, 159.76, 155.50, 153.83, 140.00, 129.09, 128.85, 126.09, 122.71, 119.71, 116.35, 113.26, 111.62, 48.94, 41.62, 31.14, 20.07, 7.96. HPLC-MS: 6.18 min, 426.2 (M+H+). HR-MS (ESI): m/z (M+H+) Calcd for C22H21O2N3ClS: 426.1038. Found: 426.1035.
(4-(2-Chlorophenoxy)-2-methylthiazol-5-yl)(4-cyclopropyl-3,4-dihydroquinoxalin-1(2H)-yl)methanone (30m)The title compound 30m was synthesized as a yellow solid in a manner similar to the title compound 30c. mp 93°C. 1H-NMR (300 MHz, CDCl3) δ: 7.31 (1H, dd, J=1.2, 7.2 Hz), 7.03–6.97 (2H, m), 6.92–6.88 (3H, m), 6.51 (1H, m), 6.33 (1H, m), 3.96 (2H, t, J=5.4 Hz), 3.46 (2H, t, J=5.4 Hz), 2.59 (3H, s), 2.32 (1H, m), 0.71 (2H, m), 0.42 (2H, m). 13C-NMR (126 MHz, CDCl3) δ: 166.37, 159.85, 155.32, 150.87, 139.77, 129.98, 127.28, 126.01, 125.68, 124.73, 124.34, 122.86, 120.07, 116.00, 113.15, 111.39, 48.83, 41.79, 31.08, 20.07, 7.90. HPLC-MS: 5.94 min, 426.2 (M+H+). HR-MS (ESI): m/z (M+H+) Calcd for C22H21O2N3ClS: 426.1038. Found: 426.1029.
(4-(4-Bromo-2,5-dichlorophenoxy)-2-methylthiazol-5-yl)(4-cyclopropyl-3,4-dihydroquinoxalin-1(2H)-yl)methanone (30n)The title compound 30n was synthesized as a light yellow solid in a manner similar to the title compound 30e. mp 128°C. 1H-NMR (300 MHz, CDCl3) δ: 7.31 (1H, dd, J=1.2, 7.2 Hz), 7.03–6.97 (2H, m), 6.92–6.88 (3H, m), 6.51 (1H, m), 6.33 (1H, m), 3.96 (2H, t, J=5.4 Hz), 3.46 (2H, t, J=5.4 Hz), 2.59 (3H, s), 2.32 (1H, m), 0.71 (2H, m), 0.42 (2H, m). 13C-NMR (126 MHz, CDCl3) δ: 166.74, 159.21, 153.82, 150.10, 139.74, 133.34, 132.81, 126.43, 125.40, 123.27, 122.79, 120.98, 116.54, 115.86, 113.13, 112.73, 48.85, 41.15, 31.12, 20.10, 7.97. HPLC-MS: 7.14 min, 538.1 (M+H+). HR-MS (ESI): m/z (M+H+) Calcd for C22H19O2N3BrCl2S: 537.9753. Found: 537.9753.
(4-(5-Chloro-2-methylphenoxy)-2-methylthiazol-5-yl)(4-cyclopropyl-3,4-dihydroquinoxalin-1(2H)-yl)methanone (31a)To a solution of 30e (102 mg, 0.2 mmol) in dry 1,4-dioxane, was added Zn(CH3)2 (333 µL, 0.4 mmol) and PdCl2(dppf)·CH2Cl2 (16.3 mg, 0.02 mmol). The reaction mixture was stirred at reflux under an atmosphere of nitrogen for 2 h. After completion, the reaction was quenched with CH3OH and stirring was continued for 10 min. The precipitate was removed by filtration and the filtrate was concentrated under reduced pressure. The residue was purified to give the title compound 31a (71 mg) as a light yellow solid in yield of 81%. mp 98°C. 1H-NMR (300 MHz, CDCl3) δ: 7.01–6.89 (4H, m), 6.83 (1H, d, J=5.7 Hz), 6.56 (1H, dt, J=1.2, 5.7 Hz), 6.05 (1H, s), 3.93 (2H, t, J=4.2 Hz), 3.42 (2H, t, J=4.2 Hz), 2.59 (3H, s), 2.27 (1H, m), 2.04 (3H, s), 0.70 (2H, m), 0.35 (2H, m). 13C-NMR (126 MHz, CDCl3) δ: 166.49, 159.81, 155.52, 153.78, 139.81, 131.31, 131.17, 126.43, 126.37, 125.83, 123.80, 122.81, 118.27, 116.26, 113.26, 111.54, 49.06, 41.37, 31.08, 20.11, 15.76, 7.83. HPLC-MS: 6.43 min, 440.2 (M+H+). HR-MS (ESI): m/z (M+H+) Calcd for C23H23O2N3ClS: 440.1194. Found: 440.1190.
(4-Cyclopropyl-3,4-dihydroquinoxalin-1(2H)-yl)(4-(2,5-dimethylphenoxy)-2-methylthiazol-5-yl)methanone (31b)The title compound 31b was prepared as a light yellow solid from 30g according to the similar procedure that preparation of 31a from 30e. mp 90°C. 1H-NMR (300 MHz, CDCl3) δ: 6.98–6.96 (3H, m), 6.92 (1H, d, J=6.0 Hz), 6.75 (1H, d, J=6.0 Hz), 6.56 (1H, m), 5.99 (1H, s), 3.94 (2H, t, J=4.2 Hz), 3.39 (2H, t, J=4.2 Hz), 2.57 (3H, s), 2.29 (1H, m), 2.17 (3H, s), 2.01 (3H, s), 0.69 (2H, m), 0.37 (2H, m). 13C-NMR (126 MHz, CDCl3) δ: 166.28, 160.28, 156.60, 153.52, 139.80, 136.27, 130.36, 126.03, 125.87, 124.88, 124.65, 122.86, 118.70, 116.18, 113.27, 110.54, 48.94, 41.77, 31.08, 20.87, 20.13, 15.76, 7.83. HPLC-MS: 6.29 min, 420.3 (M+H+). HR-MS (ESI): m/z (M+H+) Calcd for C24H26O2N3S: 420.1740. Found: 420.1732.
(4-Cyclopropyl-3,4-dihydroquinoxalin-1(2H)-yl)(4-(2,5-dichloro-4-methylphenoxy)-2-methylthiazol-5-yl)methanone (31c)The title compound 31c was prepared from 30n according to the similar procedure that preparation of 31a from 30e. mp 167°C. 1H-NMR (400 MHz, CDCl3) δ: 7.16 (1H, s), 6.97–6.87 (2H, m), 6.84 (1H, d, J=7.9 Hz), 6.58–6.49 (1H, m), 6.18 (1H, s), 3.95 (2H, t, J=5.5 Hz), 3.48 (2H, t, J=5.5 Hz), 2.58 (3H, s), 2.32 (1H, tt, J=7.1, 3.7 Hz), 2.26 (3H, s), 0.75–0.67 (2H, m), 0.45–0.37 (2H, m). 13C-NMR (126 MHz, CDCl3) δ: 166.52, 159.63, 154.87, 148.87, 139.77, 132.62, 132.29, 131.04, 126.27, 125.62, 122.84, 122.30, 120.58, 115.94, 113.13, 111.57, 48.86, 41.48, 31.07, 20.10, 19.28, 7.92. IR (KBr) cm−1: 1630.98, 1414.40, 1328.77, 1085.80, 741.82. HPLC-MS: 6.89 min, 474.2 (M+H+). HR-MS (ESI): m/z (M+H+) Calcd for C23H22O2N3Cl2S: 474.0804. Found: 474.0800.
(4-Cyclopropyl-3,4-dihydroquinoxalin-1(2H)-yl)(4-(2,5-dichloro-4-hydroxyphenoxy)-2-methylthiazol-5-yl)methanone (31d)A flask was charged with 30n (85 mg, 0.16 mmol), PdCl2(dppf)·CH2Cl2 (26 mg, 0.032 mmol), bis(pinacolato)diboron (60 mg, 0.24 mmol) and KAc (31 mg, 0.31 mmol) in 1,4-dioxane under an atmosphere of nitrogen, and then heat at 130°C for 3 h. After completion, the resulting mixture was cooled to r.t. and filtered to remove the suspended solids. The filtrate was concentrated to dryness to give a crude intermediate and that was carried forward crude into the next step. The crude intermediate was redissolved in THF, then cooled to 0°C and H2O2 (357 µL, 3.15 mmol) was added dropwise. After the addition was completed, the reaction mixture was warmed to r.t. and stirring was continued for 3 h. Upon completion, the reaction was quenched with saturated aqueous Na2SO3 and extracted with EtOAc three times. The combined organic phases were washed with brine, dried over MgSO4, filtered and evaporated under reduced pressure. The residue was purified by flash column chromatography to obtain the title compound 31b (45 mg) as a light yellow solid in 60% yield. mp 187°C. 1H-NMR (400 MHz, CDCl3) δ: 7.00–6.89 (3H, m), 6.84 (1H, d, J=7.9 Hz), 6.54 (1H, d dd, J=8.4, 6.8, 1.9 Hz), 6.11 (1H, s), 5.93 (1H, s), 3.95 (2H, t, J=5.5 Hz), 3.48 (2H, t, J=5.5 Hz), 2.59 (3H, s), 2.32 (1H, tt, J=6.8, 3.7 Hz), 0.76–0.67 (2H, m), 0.44–0.37 (2H, m). 13C-NMR (126 MHz, CDCl3) δ: 167.01, 159.65, 155.37, 148.68, 143.87, 139.98, 126.35, 125.80, 123.94, 122.81, 120.94, 118.61, 117.07, 115.93, 113.25, 110.64, 48.89, 41.57, 31.01, 19.90, 7.95. HPLC-MS: 5.28 min, 476.1 (M+H+). HR-MS (ESI): m/z (M+H+) Calcd for C22H20O3N3Cl2S: 476.0597. Found: 476.0601.
4-(5-Chloro-2-nitrophenoxy)-2-methylthiazole-5-carboxylic AcidThe carboxylic acid intermediate shown above was synthesized from 27a with the method described that preparation of 17a from 16a in yield of 93%. 1H-NMR (300 MHz, DMSO) δ: 10.28 (1H, br), 7.91 (1H, d, J=7.2 Hz), 7.15 (1H, d, J=1.2 Hz), 7.02 (1H, dd, J=1.2, 7.2 Hz), 2.54 (3H, s).
(4-(5-Chloro-2-nitrophenoxy)-2-methylthiazol-5-yl)(4-cyclopropyl-3,4-dihydroquinoxalin-1(2H)-yl)methanone (32a)The title compound 32a was obtained as a yellow solid from the intermediates shown above according to the general procedure that preparation of 13c from 12c. mp 159°C. 1H-NMR (300 MHz, CDCl3) δ: 7.83 (1H, d, J=8.7 Hz), 7.10 (1H, dd, J=2.1, 8.7 Hz), 6.93–6.81 (3H, m), 6.55–6.50 (1H, m), 6.29 (1H, s), 3.93 (2H, t, J=5.4 Hz), 3.48 (2H, t, J=5.4 Hz), 2.59 (3H, s), 2.28 (1H, m), 0.71 (2H, m), 0.38 (2H, m). 13C-NMR (126 MHz, CDCl3) δ: 166.92, 158.86, 153.11, 148.47, 139.76, 139.71, 138.51, 126.38, 126.06, 125.27, 124.06, 122.91, 121.36, 115.91, 113.47, 113.00, 48.81, 41.13, 31.16, 20.05, 7.95. IR (KBr) cm−1: 1629.14, 1600.68, 1528.36, 1503.64, 1410.83, 1331.04, 1229.17, 756.33. HPLC-MS: 6.02 min, 471.2 (M+H+). HR-MS (ESI): m/z (M+H+) Calcd for C22H20O4N4ClS: 471.0888. Found: 471.0875.
(4-Cyclopropyl-3,4-dihydroquinoxalin-1(2H)-yl)(2-methyl-4-(5-methyl-2-nitrophenoxy)thiazol-5-yl)methanone (32b)The title compound 32b was obtained as a light yellow solid according to the procedure that preparation of the title compound 32a. mp 94°C. 1H-NMR (300 MHz, CDCl3) δ: 7.81 (1H, d, J=6.3 Hz), 6.97–6.88 (4H, m), 6.54 (1H, m), 6.16 (1H, s), 3.95 (2H, t, J=4.2 Hz), 3.47 (2H, t, J=4.2 Hz), 2.56 (3H, s), 2.28 (4H, m), 0.70 (2H, m), 0.38 (2H, m). 13C-NMR (126 MHz, CDCl3) δ: 166.63, 163.28, 159.39, 154.38, 147.97, 145.65, 139.78, 138.10, 127.08, 125.92, 125.21, 124.82, 123.10, 121.75, 115.86, 113.07, 48.84, 41.70, 31.13, 21.45, 20.03, 7.91. HPLC-MS: 5.72 min, 451.2 (M+H+). HR-MS (ESI): m/z (M+H+) Calcd for C23H23O4N4S: 451.1435. Found: 451.1430.
(4-Cyclopropyl-3,4-dihydroquinoxalin-1(2H)-yl)(4-(2,5-dichlorophenoxy)-2-ethylthiazol-5-yl)methanone (37a)The title compound 37a was obtained as a light yellow solid according to the procedure that preparation of the title compound 30c. mp 102°C. 1H-NMR (400 MHz, CDCl3) δ: 7.20 (1H, d, J=8.5 Hz), 6.95–6.87 (2H, m), 6.87–6.82 (1H, m), 6.79 (1H, d, J=7.8 Hz), 6.52 (1H, td, J=8.1, 7.6, 1.5 Hz), 6.14 (1H, s), 3.94 (2H, t, J=5.5 Hz), 3.49 (2H, t, J=5.4 Hz), 2.91 (2H, q, J=7.5 Hz), 2.34–2.27 (1H, m), 1.33 (3H, t, J=7.6 Hz), 0.75–0.69 (2H, m), 0.46–0.40 (2H, m). 13C-NMR (126 MHz, CDCl3) δ: 173.34, 159.55, 153.98, 150.97, 139.63, 132.40, 130.24, 126.32, 125.33, 124.37, 122.78, 122.23, 119.64, 115.85, 113.06, 112.40, 48.82, 41.26, 31.12, 27.58, 13.45, 7.91. IR (KBr) cm−1: 1632.63, 1475.48, 1414.25, 1329.43, 734.26. HPLC-MS: 7.33 min, 474.1 (M+H+). HR-MS (ESI): m/z (M+H+) Calcd for C23H22O2N3Cl2S: 474.0804. Found: 474.0797.
(4-(3-Chlorophenoxy)-2-ethylthiazol-5-yl)(4-cyclopropyl-3,4-dihydroquinoxalin-1(2H)-yl)methanone (37b)The title compound 37b was obtained as a light yellow solid according to the procedure that preparation of the title compound 30i. mp 84°C. 1H-NMR (400 MHz, CDCl3) δ: 7.10 (1H, t, J=8.1 Hz), 7.02–6.93 (3H, m), 6.81 (1H, d, J=8.0 Hz), 6.55–6.47 (3H, m), 3.92 (2H, t, J=5.6 Hz), 3.37 (2H, t, J=5.6 Hz), 2.91 (2H, q, J=7.5 Hz), 2.35–2.28 (1H, m), 1.33 (3H, t, J=7.6 Hz), 0.78–0.72 (2H, m), 0.47–0.41 (2H, m). 13C-NMR (126 MHz, CDCl3) δ: 173.23, 159.77, 155.85, 154.73, 139.87, 134.35, 129.78, 126.18, 125.81, 123.69, 122.67, 118.47, 116.33, 116.06, 113.20, 112.24, 48.94, 41.55, 31.19, 29.70, 27.58, 13.47, 7.94. HPLC-MS: 7.02 min, 440.2 (M+H+). HR-MS (ESI): m/z (M+H+) Calcd for C23H23O2N3ClS: 440.1194. Found: 440.1188.
(4-Cyclopropyl-3,4-dihydroquinoxalin-1(2H)-yl)(4-(2,5-dichlorophenoxy)-2-isopropylthiazol-5-yl)methanone (37c)The title compound 37c was obtained as a light yellow solid according to the procedure that preparation of the title compound 30c. mp 89°C. 1H-NMR (400 MHz, CDCl3) δ: 7.20 (1H, d, J=8.5 Hz), 6.92 (1H, dd, J=8.5, 2.3 Hz), 6.90–6.82 (2H, m), 6.79 (1H, d, J=7.8 Hz), 6.51 (1H, ddd, J=8.1, 7.0, 1.8 Hz), 6.17 (1H, s), 3.95 (2H, t, J=5.4 Hz), 3.49 (2H, t, J=5.5 Hz), 3.17 (1H, p, J=6.9 Hz), 2.34–2.27 (1H, m), 1.33 (6H, d, J=6.9 Hz), 0.76–0.68 (2H, m), 0.48–0.43 (2H, m). 13C-NMR (126 MHz, CDCl3) δ: 178.20, 159.69, 153.79, 151.00, 139.58, 132.34, 130.22, 126.26, 125.26, 124.21, 122.76, 122.14, 119.51, 115.84, 113.05, 112.15, 48.79, 41.32, 33.82, 31.16, 29.70, 22.49, 14.13, 7.93. HPLC-MS: 7.74 min, 488.2 (M+H+). HR-MS (ESI): m/z (M+H+) Calcd for C24H24O2N3Cl2S: 488.0961. Found: 488.0951.
(4-(3-Chlorophenoxy)-2-isopropylthiazol-5-yl)(4-cyclopropyl-3,4-dihydroquinoxalin-1(2H)-yl)methanone (37d)The title compound 37d was obtained as a light yellow solid according to the procedure that preparation of the title compound 30i. mp 81°C. 1H-NMR (400 MHz, CDCl3) δ: 7.12–7.06 (1H, m), 7.02–6.92 (3H, m), 6.81 (1H, d, J=7.8 Hz), 6.55–6.49 (3H, m), 3.92 (2H, t, J=5.6 Hz), 3.36 (2H, t, J=5.5 Hz), 3.17 (1H, p, J=6.9 Hz), 2.35–2.28 (1H, m), 1.34 (6H, d, J=6.9 Hz), 0.79–0.72 (2H, m), 0.49–0.44 (2H, m). 13C-NMR (126 MHz, CDCl3) δ: 178.12, 159.89, 155.93, 154.48, 139.81, 134.30, 129.73, 126.14, 125.73, 123.52, 122.63, 118.33, 116.34, 115.86, 113.19, 112.14, 48.90, 41.64, 33.82, 31.24, 29.70, 22.50, 7.95. HPLC-MS: 7.42 min, 454.2 (M+H+). HR-MS (ESI): m/z (M+H+) Calcd for C24H25O2N3ClS: 454.1351. Found: 454.1349.
(4-Cyclopropyl-3,4-dihydroquinoxalin-1(2H)-yl)(4-(2,5-dichlorophenoxy)-2-phenylthiazol-5-yl)methanone (37e)The title compound 37e was obtained as a yellow solid according to the procedure that preparation of the title compound 30c. mp 136°C. 1H-NMR (400 MHz, CDCl3) δ: 7.84–7.78 (2H, m), 7.47–7.37 (3H, m), 7.24 (1H, d, J=8.6 Hz), 6.99–6.81 (4H, m), 6.55–6.47 (1H, m), 6.27 (1H, s), 3.98 (2H, t, J=5.5 Hz), 3.52 (2H, t, J=5.5 Hz), 2.36–2.29 (1H, m), 0.77–0.69 (2H, m), 0.49–0.42 (2H, m). 13C-NMR (126 MHz, CDCl3) δ: 166.87, 159.42, 155.20, 150.94, 139.69, 132.54, 132.37, 131.11, 130.22, 129.01, 126.41, 126.17, 125.38, 124.55, 122.73, 122.53, 120.07, 115.97, 113.10, 112.96, 48.82, 41.25, 31.14, 7.95. IR (KBr) cm−1: 1639.69, 1472.36, 1407.45, 1336.07, 731.61. HPLC-MS: 8.15 min, 522.2 (M+H+). HR-MS (ESI): m/z (M+H+) Calcd for C27H22O2N3Cl2S: 522.0804. Found: 522.0800.
(4-(3-Chlorophenoxy)-2-phenylthiazol-5-yl)(4-cyclopropyl-3,4-dihydroquinoxalin-1(2H)-yl)methanone (37f)The title compound 37f was obtained as a yellow solid according to the procedure that preparation of the title compound 30i. mp 123°C. 1H-NMR (400 MHz, CDCl3) δ: 7.87–7.80 (2H, m), 7.47–7.36 (3H, m), 7.12 (1H, t, J=7.9 Hz), 7.06–6.93 (3H, m), 6.86 (1H, d, J=7.9 Hz), 6.59 (2H, d, J=8.4 Hz), 6.56–6.48 (1H, m), 3.97–3.94 (2H, m), 3.40 (2H, t, J=5.6 Hz), 2.38–2.30 (1H, m), 0.81–0.71 (2H, m), 0.51–0.43 (2H, m). 13C-NMR (126 MHz, CDCl3) δ: 166.81, 159.60, 155.85, 155.77, 139.92, 134.29, 132.61, 131.04, 129.72, 128.98, 126.28, 126.13, 125.85, 123.80, 122.58, 118.75, 116.44, 116.33, 113.24, 112.95, 48.94, 41.57, 31.22, 7.97. HPLC-MS: 7.05 min, 488.2 (M+H+). HR-MS (ESI): m/z (M+H+) Calcd for C27H23O2N3ClS: 488.1194. Found: 488.1186.
(4-Cyclopropyl-3,4-dihydroquinoxalin-1(2H)-yl)(4-(2,5-dichlorophenoxy)-2-(pyridin-2-yl)thiazol-5-yl)methanone (37g)The title compound 37g was obtained as a yellow solid according to the procedure that preparation of the title compound 30c. mp 169°C. 1H-NMR (400 MHz, CDCl3) δ: 8.60 (1H, d, J=5.0 Hz), 7.90 (1H, d, J=7.9 Hz), 7.72 (1H, t, J=7.9 Hz), 7.40–7.18 (2H, m), 7.03–6.77 (4H, m), 6.49 (1H, t, J=7.5 Hz), 6.21 (1H, s), 3.98 (2H, d, J=5.7 Hz), 3.52 (2H, t, J=5.4 Hz), 2.32 (1H, s), 0.73 (2H, d, J=6.5 Hz), 0.44 (2H, s). 13C-NMR (126 MHz, CDCl3) δ: 167.27, 159.52, 155.16, 150.98, 150.06, 149.55, 139.70, 137.05, 132.38, 130.24, 126.46, 125.31, 125.24, 124.48, 122.88, 122.46, 119.88, 119.79, 115.91, 115.72, 113.09, 48.81, 41.13, 31.12, 7.93. IR (KBr) cm−1: 1627.25, 1471.82, 1398.62, 1230.21, 1075.35, 738.43. HPLC-MS: 7.44 min, 523.2 (M+H+). HR-MS (ESI): m/z (M+H+) Calcd for C26H21O2N4Cl2S: 523.0757. Found: 523.0747.
(4-(3-Chlorophenoxy)-2-(pyridin-2-yl)thiazol-5-yl)(4-cyclopropyl-3,4-dihydroquinoxalin-1(2H)-yl)methanone (37h)The title compound 37h was obtained as a yellow solid according to the procedure that preparation of the title compound 30i. mp 142°C. 1H-NMR (400 MHz, CDCl3) δ: 8.60 (1H, dt, J=4.8, 1.3 Hz), 7.95–7.91 (1H, m), 7.72 (1H, td, J=7.8, 1.8 Hz), 7.36–7.30 (1H, m), 7.13 (1H, t, J=8.0 Hz), 7.05–6.93 (3H, m), 6.83 (1H, d, J=7.9 Hz), 6.58 (2H, d, J=10.2 Hz), 6.50 (1H, td, J=8.0, 7.3, 1.9 Hz), 3.96 (2H, t, J=5.6 Hz), 3.40 (2H, t, J=5.6 Hz), 2.38–2.30 (1H, m), 0.79–0.72 (2H, m), 0.49–0.42 (2H, m). 13C-NMR (126 MHz, CDCl3) δ: 167.18, 159.72, 155.86, 150.16, 149.55, 139.93, 137.04, 134.34, 130.01, 129.77, 126.33, 125.70, 125.25, 123.78, 122.75, 119.75, 118.67, 116.37, 116.23, 115.68, 113.23, 48.92, 41.45, 31.21, 7.95. HPLC-MS: 7.26 min, 489.2 (M+H+). HR-MS (ESI): m/z (M+H+) Calcd for C26H22O2N4ClS: 489.1147. Found: 489.1137.