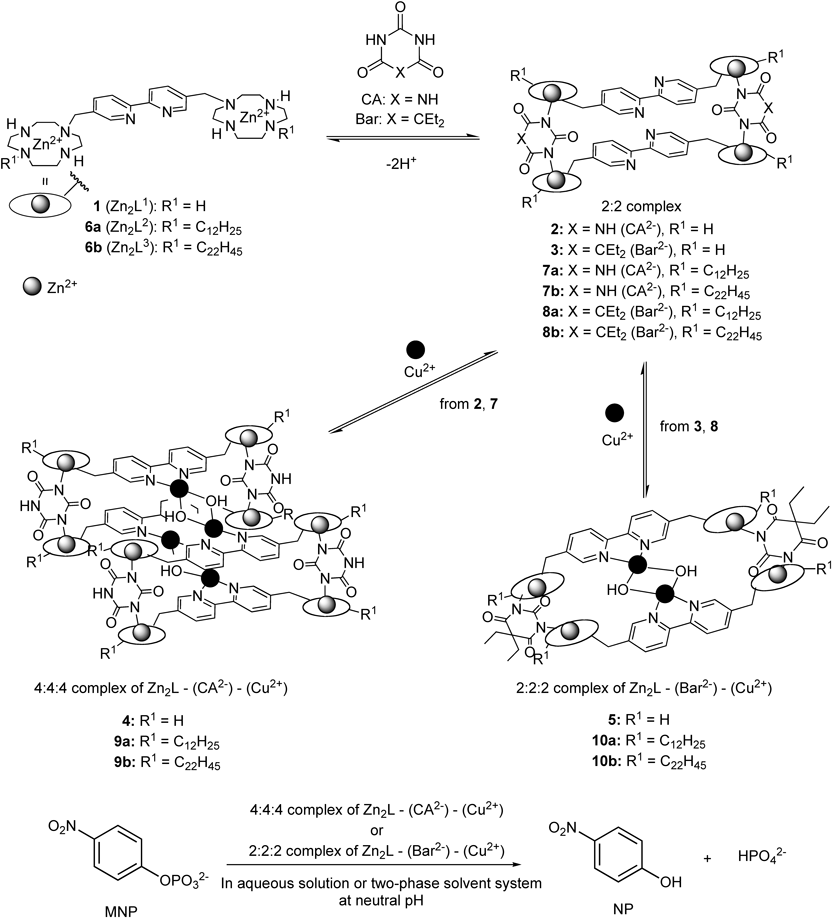
Results and Discussion
Synthesis of Dimeric Zn2+ Complexes Containing Long Alkyl ChainsHydrophobic bis(Zn2+–cyclen)bpy containing long alkyl chains 6a (Zn2L2) and 6b (Zn2L3) were synthesized as shown in Chart 3. Cyclen (1,4,7,10-tetraazacyclododecane) was reacted with tert-butoxycarbonyl (Boc)-OSu to give 2Boc-cyclen 13 following to a previously reported procedure.61) The monoalkylation of 13 with alkyl (dodecyl or docosyl) iodide 15a and b, prepared from 14a and b, afforded 16a and b. The reaction of 5,5′-bis(bromomethyl)-2,2′-bipyridine 1736,46,47,62) with 16a and b gave 18a and b, the Boc groups of which were removed by treatment with aqueous HBr to yield 19a and b as HBr salts. After deprotonation, the free form of 19a was reacted with 2 eq of Zn(NO3)2·6H2O to give the dimeric zinc(II) complex, 6a (Zn2L2). The free form of 19b was also reacted with 2 equiv. of Zn(ClO4)2·6H2O to give 6b (Zn2L3). As hydrophobic barbital blocks, 20, 22 and 23 were prepared according to our paper47) and 25 having 4-trifluoromethylbenzyl group was prepared as shown in Chart 4.63,64)

Chart 3. Synthesis of 6a (Zn2L2) and 6b (Zn2L3)

Chart 4. Functionalized Barbital Derivatives 20–25 and Synthesis of 25
UV/Vis titrations of 6a (80 µM) with CA and Bar were carried out in a homogeneous mixture of n-butanol–50 mM 2-(4-(2-hydroxymethyl)-1-piperazinyl) ethanesulfonic acid (HEPES) buffer (pH 7.4 with I=0.1 (NaNO3)) (9 : 1), because 6a (the building block of 9a, 10a) is soluble in n-butanol, but not in H2O and CHCl3. As shown in Fig. 1a, 6a (80 µM) has an absorption maxima (λmax) at 292 nm, which decreased upon the addition of CA reaching a plateau at a 1 : 1 ratio, as shown in the inset. The change in the spectrum of 6a upon the addition of Bar is also shown in Fig. 1b. The absorbance at 292 nm increased upon the addition of Bar2− and reaching a plateau at a 1 : 1 ratio, as shown in the inset. These results suggest that the 2 : 2 complexes, 7a and 8a, are formed from 6a with CA2− and Bar2−, respectively.
Figure 2 shows the results of UV/Vis titrations of 7a and 8a (40 µM) with Cu(NO3)2, which exhibit a red-shift in the absorption maxima from ca. 290 nm to ca. 308 nm. The absorption at 293 nm and 290 nm for 7a and 8a, reached a plateau at [Cu2+]/[2 : 2 complex]=2.0, as indicated in the insets of Fig. 2. These results suggest that 9a and 10a are quantitatively formed at µM order concentrations, as was reported in our previous publications on the formation of 4 and 5.46,47)
Complexation Behaviors of 6b with CA2− or Bar2− and Cu2+ in Two-Phase Systems (CHCl3–H2O), as Studied by UV/Vis TitrationUV/Vis titrations of 6a with CA or Bar in a two-phase solvent system (CHCl3–50 mM HEPES buffer (pH 7.4 with I=0.1 (NaNO3)) (9 : 1)) were unsuccessful due to the low solubility of 6a in both CHCl3 and H2O. Thus, UV/Vis titrations of 6b (20 µM) with CA or Bar were carried out in the same two-phase solvent system (Fig. 3). 6b (20 µM) has an absorption maxima (λmax) at 297 nm, which decreased upon the addition of CA2−, reaching a plateau at a 1 : 1 ratio, as shown in the inset. The spectral changes upon the addition of Bar are also shown in Fig. 3b, in which the absorption at 302 nm decreased upon the addition of Bar2− and reached a plateau at a 1 : 1 ratio, as shown in the inset. These results suggest that 6b forms the 2 : 2 complexes 7b and 8b with CA and Bar, respectively, in a manner similar to that for the formation of 7a and 8a (Chart 1, Fig. 1).
UV/Vis titrations of 7b or 8b (20 µM) with Cu(ClO4)2 in a two-phase solvent system (CHCl3–50 mM HEPES buffer (pH 7.4 with I=0.1 (NaNO3)) were also conducted. Because the spectral changes occur very slowly, UV/Vis spectra (of CHCl3 phase) in the presence of Cu2+ (0–4 eq) were measured after incubation with Cu2+ for ca. 24 h at 37°C in CHCl3–50 mM HEPES buffer (pH 7.4 with I=0.1 (NaNO3)) (2/8). As shown in Fig. 4, the UV/Vis spectra of 7b and 8b in the CHCl3 phase exhibited a red-shift from ca. 287 nm to ca. 304 nm, indicating the complexation of the 2 : 2 complex of 7b and 8b with Cu2+ in the two-phase solvent system, which reached a plateau at [Cu2+]/[2 : 2 complex]=2.0 (insets of Fig. 4), indicative of the quantitative formation of 9b and 10b at µM order concentrations (Note that the concentrations of 9 and 10 in the two-phase solvent system are defined by their concentrations with reference to the total volume of solvent including CHCl3 and H2O).
Partition Properties of MNP, BNP and NP between Organic Phase and Aqueous PhaseBefore the hydrolysis experiment, the distribution of MNP and NP between the aqueous and organic (CHCl3) phases was measured in the presence and absence of 10b. A solution of MNP (100 µM in the initial aqueous phase, before the addition of CHCl3) in a buffer solution (50 mM HEPES buffer at pH 7.4 with I=0.1 (NaNO3)) (2.4 mL) and CHCl3 (0.6 mL) were mixed and stirred vigorously for 15 min at 25°C. This turbid mixture was centrifuged (3000 rpm×10 min at 25°C) to obtain two cleanly separated phases. The extraction rates of MNP into the organic phase in the absence and presence of 10b (20 µM in the total solution) were determined to be ca. 3% and ca. 8%, respectively, based on UV/Vis absorption spectra of the aqueous layer (Fig. 5a). On the other hand, the extraction rates of BNP (100 µM) into the organic phase in the absence and presence of 10b (20 µM in total solution) were determined to be ca. 1% and ca. 14%, respectively, indicating that BNP is extracted into CHCl3 in the presence of 10b more effectively than MNP (Fig. 5b).
The NP distribution of the initial concentration was similarly measured (100 µM in 50 mM HEPES buffer at pH 7.4 with I=0.1 (NaNO3) (4 mL) and CHCl3 (1 mL)), and the partition ratio of NP was determined to be 79% in H2O at pH 7.4.
The Hydrolysis of MNP by Hydrophobic 4 : 4 : 4 Complexes and 2 : 2 : 2 Complexes in Single and Two-Phase Solvent SystemsThe hydrolysis of MNP (100 µM) was carried out in the presence of 9a or 10a (100 µM in the total amount of solvent) in n-butanol–50 mM HEPES buffer (pH 7.4, with I=0.1 (NaNO3)) (8 : 2) at 37°C. All the hydrolysis experiments in this study are performed by shaking vigorously using shaking water bath (Tokyo Glass Kikai, FWB-1) (shaking speed: 196 rpm), equipped with temperature-controlling unit (Nissin Rika, NC-301D). Figure 6 shows that 9a and 10a promote the hydrolysis of MNP, the yield of which reached a plateau of ca. 4% after 3 d. The results for the hydrolysis of MNP by 4 and 5 in a single solvent (H2O)46,47) and a two-phase solvent system (n-butanol–H2O) are compared in Fig. 6. It is obvious that the hydrolysis of MNP in n-butanol–H2O is much slower than that in H2O alone. Since n-butanol is somewhat soluble in H2O (9.5 mL n-butanol–100 mL H2O), we assume that HPO42− is transferred to the n-butanol phase and inhibits the activity by 9a and 10a stronger than in H2O alone.
The hydrolysis of MNP (100 µM) was next carried out in CHCl3–50 mM HEPES buffer (pH 7.4) with I=0.1 (NaNO3) (2 : 8) in the presence of 9b or 10b (20 µM or 100 µM in total solution) at 37°C (Figs. 7, 8). While the two supermolecules 9b or 10b ([9b] or [10b]=100 µM in the total solution) promote MNP hydrolysis, their initial rates were found to be slower than those by 4 and 5 ([4] or [5]=100 µM) in a single aqueous solution (Fig. 7). In contrast, 9b or 10b ([9b] or [10b]=20 µM in total solution) in a two-phase solvent system exhibited a higher hydrolytic activity for MNP than that of 5 (20 µM) in aqueous solution (Fig. 8), because the stability of the catalytic amount of 5 (20 µM) in the aqueous solution is low.47,65,66) In both reaction conditions (Figs. 7, 8), the activity of 10b per Cu2(μ-OH)2 site is somewhat higher than that of 9b, assuming that the two Cu2(μ-OH)2 centers of 9b function independently. The acceleration in the hydrolysis of MNP in the presence of 6b, 6b+Bar (1 : 1), 6b+CA (1 : 1) and Cu2+ was negligible under the same conditions (Fig. 8). Although 6b+Cu2+ (1 : 1) facilitated the hydrolysis of MNP, the hydrolysis yield (ca. 5%) in 7 d was almost a half of that of 10b (ca. 12%).67) This can be attributed to the lower stability of 6b+Cu2+ complex in CHCl3–H2O than that of 10b. These collective results support the conclusion that all of the components contained in 6b, CA or Bar, and Cu2+ are required for the hydrolysis of MNP.
Moreover, we tested the hydrolysis of MNP (100 µM in total solution) by 10b (20 µM in total solution) in various two-phase solvent systems such as 1,2-dichloroethane (DCE)–50 mM HEPES buffer (pH 7.4) (2 : 8), DCE–CHCl3–50 mM HEPES buffer (pH 7.4) (1 : 1 : 8), hexanes–CHCl3–50 mM HEPES buffer (pH 7.4) (0.7 : 1.3 : 8.0), and CHCl3–MeOH–50 mM HEPES buffer (pH 7.4) (2 : 1 : 7). However, the activity of 10b for hydrolysis in these solvent systems was essentially the same or lower than that in CHCl3–50 mM HEPES buffer (pH 7.4) (data not shown).
In our previous paper, we reported that functional groups on their side chains of barbital units affect the hydrolytic activity of 2 : 2 : 2 supermolecules in aqueous solution.47) Therefore, we examined the effect of the barbital derivatives 20–25 on the MNP hydrolytic activity of the corresponding 2 : 2 : 2 complexes 10c–h in a two-phase solvent system (Fig. 9). A lower activity was observed for 10c, d, f, and g than that of 10b, as shown in Fig. 9.
Cozzi et al. reported the aromatic interactions between two benzene rings having electron-withdrawing groups such as nitro or ester groups.68) Therefore, we decided to use 22 and 25 as barbital analogues having benzyloxyethyl side chains, expecting the more strong complexation of MNP with supermolecules and more efficient extraction of MNP to organic layer (Fig. 9) (we could not synthesize the corresponding (4-nitrobenzyloxy)ethyl derivative). It was expected that 4-trifluoromethylbenzyl group of 10h is located near to the Cu2(μ-OH)2 site and providing hydrophobic environment for the substrate in comparison with 10b. Interestingly, it was disclosed that 10e (formed from 6a+22+Cu2+) and 10h have higher hydrolysis activity than 10b, as displayed in Fig. 9.
Moreover, the effect of the concentration of MNP (1000 µM) on hydrolysis by 9b, 10b and h was examined. As shown in Fig. 10, reactions by 9b, 10b and h became slower after the production of ca. 1 eq of HPO42− against Cu2(μ-OH)2 centers of supermolecules, and the reactions reached a plateau with yields of ca. 2–4% in 3–4 d, possibly due to product inhibition by HPO42−.
MNP hydrolysis by 10b and h was carried out at the increasing concentration of [MNP] (100–1000 µM in total solvent system including CHCl3 and H2O) for 24 h at 37°C. Interestingly, 10h is more active than 10b at [MNP]=100 µM, but both complexes have almost same hydrolytic activity at [MNP]>500 µM as summarized in Fig. 11a (x axis implies the concentration of MNP and, at the same time, it implies [MNP]/[supermolecule] ratio). This finding has suggested that the catalytic site of 10b and h are partially occupied by the substrate at [MNP]<500 µM and fully occupied at higher concentrations.
This information has prompted us to check whether the MNP hydrolysis follows Michaelis–Menten kinetics. In Lineweaver–Burk plot (Fig. 11b), concentrations of substrate (MNP) and produced NP in the total solution of the two-phase system are used for calculation (V0=initial hydrolysis rate). From this graph, approximate Vmax (the maximum rate in the NP production from MNP by 10b) and Km (Michaelis constant) values for 10b were determined to be (1.4±0.4)×10−2 µM/min and (5.4±0.5)×102 µM, respectively. As shown in Table 1, the Km value for 10b is in fairly good agreement with that of 5 in the single aqueous solution, although Vmax of 10b is ca. 1/6 as small as that of 5.47) On the other hand, Km value for 10h ((53±4) µM) is ca. 10 times smaller than that of 10b (Vmax for 10h is (1.0±0.3)×10−2 µM/min). Therefore, the increase in hydrolysis rate of MNP by 10h can be attributed to lower Km value of 10h than that of 10b, which could be explained by hydrophobic interaction and/or interactions between electron-deficient moieties of the supermolecules and the substrate.
Table 1. Kinetic Parameters (
Vmax and
Km) for Hydrolysis of MNP by
10b and
h in Comparison with
5 | Vmax (µM/min) | Km (µM) |
|---|
| 5a) | (8.9±0.2)×10−2 b) | (4.1±0.3)×102 b) |
| 10bc) | (1.4±0.4)×10−2 | (5.4±0.5)×102 |
| 10hc) | (1.0±0.3)×10−2 | 53±4 |
a) From ref. 47. b) Determined in a single aqueous solution (10 mM HEPES buffer, pH 7.4). c) In CHCl3–50 mM HEPES buffer (pH 7.4).
Our speculation regarding the product inhibition of MNP hydrolysis in two-phase solvent system is shown in Chart 5. We initially expected that the inorganic phosphate would be released from the Cu2(μ-OH)2 sites of hydrophobic supermolecules at the organic–water interface. However, the supermolecules used in this work may have been too hydrophobic and hence their catalytic Cu2(μ-OH)2 sites are located too far from the interface (from water),69) thus preventing an effective exchange of HPO42− and MNP. Furthermore, the possibility that a stable intermediate 29 is formed, in which one of oxyanions of inorganic phosphate bridges two Cu2+ ions, thus slowing the release of HPO42−. At the same time, hydrophobicity of supramolecular complexes is important to extract MNP to organic layer more effectively, giving more stable supermolecule–substrate complexes (smaller Km).69)

Chart 5. Speculation about Hydrolysis Acceleration and Product Inhibition in Two-Phase Solvent System
Next, the hydrolysis of phosphodiester BNP by the hydrophobic supermolecules 9b, 10b, and h was carried out in two-phase solvent systems (Chart 6). Although there are various artificial metal complexes that promote BNP hydrolysis,48–60) complexes that function as catalysts remain limited.49–51) The hydrolytic activity of 10b (20 µM) for BNP (100 µM) was examined in CHCl3–50 mM HEPES buffer (pH 7.4) with I=0.1 (NaNO3) (2 : 8) at 37°C.

Chart 6. BNP Hydrolysis by 10b in a Two-Phase Solvent System
As shown in Fig. 12a, 10b accelerates the hydrolysis of BNP with ca. 35% yield (the catalytic turnover number (CTN) is ca. 1.6) in 10 d in a two-phase solvent system, indicating its catalytic activity in BNP hydrolysis. The hydrolytic activity of 9b was ca. 15% in 10 d and CTN is estimated to be 0.4, assuming that the two Cu2(μ-OH)2 centers function independently. These findings are in contrast to previous data for hydrophilic supermolecules 4 and 5, for which the hydrolysis of BNP and tris(4-nitrophenyl)phosphate (TNP) in an aqueous solution was accelerated to only a negligible extent.46,47) Since the BNP monoanion interacts with the Cu2(μ-OH)2 centers of 10b in CHCl3 more strongly than in an aqueous solution, the use of a two-phase solvent system (CHCl3–H2O) is necessary for the efficient hydrolysis of BNP by 10b. The negligible hydrolytic activity of the 2 : 2 complex 8b (6b (20 µM)+Bar (20 µM)) or Cu2+ (40 µM) was observed under the same reaction conditions (Fig. 12b). Although the initial rate of BNP hydrolysis by hydrophobic Zn2L 6b (40 µM) is greater than that by 10b (20 µM), CTN is ca. 0.4 (based on the assumption that two Zn2+-cyclen units in 6b function independently), possibly because the produced MNP binds strongly to the Zn2+ in 6b and does not undergo hydrolysis.48) As shown in Fig. 12a, higher hydrolytic activity of 10e (ca. 42% and CTN is ca. 2.1 in 10 d) and 10h (ca. 45% and CTN is ca. 2.3 in 10 d) than that of 10b is also observed in hydrolysis of BNP.
Figure 13 displays the catalytic activity of 10b in the presence of a large excess of BNP ([BNP]=3.0 mM, 150 fold over [10b]) (ca. 9% in 6 d, CTN: 13). The yields for 9b and 6b+Cu2+ were ca. 3.5% (CTN: 3.5 against each Cu2(μ-OH)2 center) and ca. 4% in 6 d, which are lower than that for 10b, and the yield for 6b (40 µM) is ca. 0.7% due to the strong product inhibition. Hydrolysis reactions were negligibly accelerated by Cu2+ or 8b (6b (40 µM)+Bar (40 µM)) under the same conditions. These results strongly support the conclusion that the supramolecular system formed from the self-assembly of the three components (6b, Bar, and Cu2+) is required for catalytic hydrolysis of BNP in a two-phase solvent system. As shown in Fig. 13, almost the same hydrolytic activity of 10b, e, and 10d for BNP was observed at high BNP concentration ([BNP]=3.0 mM), while 10e and h exhibit higher hydrolytic activity than 10b at [BNP]=100 µM (Fig. 12a). This behavior is similar to the hydrolysis of MNP by hydrophobic supermolecules (Figs. 10, 11).
As shown in Fig. 13, the hydrolysis reactions of BNP by 10b and h became slower as reaction proceeded, probably due to the product inhibition. This result suggests that HPO42− production inhibits the catalytic cycle of 10b since MNP produced by hydrolysis of BNP is also hydrolyzed by 10b (Chart 6). Indeed, the product inhibition by HPO42− is supported by the concentration-dependent inhibition of HPO42− for the hydrolysis of BNP by 10b, as summarized in Fig. 14. Moreover, we tried to measure 31P-NMR spectra of sample solution used for hydrolysis reaction of BNP (3.0 mM) in the presence of 10b (20 µM) in CHCl3–H2O (reaction conditions: 37°C for 6 d). However, 31P signal of HPO42− was not observed in the aqueous layer of sample solution, while 31P signals of BNP and MNP were observed. On the other hand, the 31P signals of phosphates in organic layer were negligible, possibly due to the coordination of phosphates to the Cu2(μ-OH)2 core of 10b.
Experimental
General InformationAll reagents and solvents were of the highest commercial quality and were used without further purification, unless otherwise noted. N,N-Dimethylformamide (DMF) was obtained by distillation from calcium hydride. Tetrahydrofuran (THF) was obtained by distillation from Na/benzophenone. MNP and BNP were purchased from Nacalai Tesque (Japan). Compound 21 was purchased from TCI Co., Ltd. (Japan). Sodium salt of compound 24 was purchased from Wako Pure Chemical Industries, Ltd. (Japan). All aqueous solutions were prepared using deionized and distilled water. The Good’s buffer reagents (Dojindo, pKa at 20°C) were obtained from commercial sources: HEPES (2-(4-(2-hydroxyethyl)-1-piperazinyl))ethanesulfonic acid, pKa=7.6). UV/Vis spectra were recorded on a JASCO V-550 spectrophotometer with quartz cuvettes (path length: 10 mm or 2 mm). IR spectra were recorded on a Perkin-Elmer attenuated total reflectance (ATR)-IR spectrometer 100 at room temperature. Melting points were measured by a Yanaco MP-J3 Micro Melting Point apparatus and are uncorrected. 1H- (300 MHz) and 13C- (75 MHz) NMR spectra at 25±0.1°C were recorded on a JEOL Always 300 spectrometer. 1H-NMR (400 MHz), 19F-NMR (376 MHz), and 31P-NMR (162 MHz) were recorded on a JEOL Lambda 400 spectrometer. Tetramethylsilane (TMS) was used as internal reference for 1H- and 13C-NMR measurements in CDCl3, DMSO-d6 or CD3OD. Trifluoroacetic acid (TFA) was used as external reference (δ: −76.5 ppm) for 19F-NMR measurements. Eighty five percent H3PO4 was used as an external reference (δ: 0.00 ppm) for the 31P-NMR measurements. Mass spectra were recorded on a JEOL JMS-SX102A and Varian 910-MS. Elemental analyses were performed on a Perkin-Elmer CHN 2400 series II CHNS/O analyzer at the Research Institute for Science and Technology, Tokyo University of Science. TLC and silica gel column chromatographies were performed using Merck Silica gel 60 F254 TLC plate or Fuji Silysia Chemical CHROMATOREX NH-TLC PLATE, and Fuji Silysia Chemical FL-100D or Fuji Silysia Chemical CHROMATOREX NH Chromatography Silica Gel, respectively.
Synthesis1-Dodecyl-4,10-bis(tert-butyloxycarbonyl)-1,4,7,10-tetraazacyclododecane (16a)A mixture of 1-bromododecane 14a (493 mg, 1.98 mmol) and sodium iodide (296 mg, 1.98 mmol) in acetone (5 mL) was heated at reflux for 5 h under argon atmosphere. After the reaction, the mixture was cooled to room temperature, the insoluble compounds were filtered off and washed with diethyl ether (3 mL). The filtrate was concentrated under reduced pressure to give a pale yellow oil 15a (586 mg), which was used in the next step without further purification. To a mixture of 1,7-bis(tert-butyloxycarbonyl)-1,4,7,10-tetraazacyclododecane 1361) (669 mg, 1.80 mmol) and potassium carbonate (248 mg, 1.80 mmol) in MeCN (30 mL), a solution of 15a (586 mg, 1.98 mmol) in MeCN (15 mL) was slowly added over a period of 15 min. The mixture was stirred at room temperature for 72 h under argon atmosphere. The reaction mixture was evaporated under reduced pressure, the residue suspended in H2O (40 mL), and the solution extracted with CHCl3 (40 mL×3). The organic layer was washed with brine, dried over Na2SO4, filtered and concentrated under reduced pressure. The remaining residue was purified by NH silica gel column chromatography (hexane–CHCl3=1 : 1) to give 16a as a colorless oil (321 mg, 33% yield). IR (ATR) cm−1: 3223, 2923, 2853, 1692, 1458, 1405, 1364, 1245, 1158, 772. 1H-NMR (300 MHz, CDCl3–TMS) δ: 3.40–3.20 (m, 8H), 2.78–2.66 (m, 8H), 2.55–2.45 (m, 2H), 1.45 (s, 18H), 1.25 (m, 20H), 0.88 (t, 3H, J=6.9 Hz). 13C-NMR (75 MHz, CDCl3–TMS) δ: 156.1, 155.9, 79.6, 79.5, 54.4, 53.8, 53.1, 51.1, 50.4, 50.2, 50.0, 49.9, 49.6, 49.3, 48.8, 48.5, 48.1, 47.9, 47.6, 29.8, 29.7, 29.6, 29.4, 28.5, 27.8, 25.0, 22.8. FAB-MS m/z: 541.4693 (Calcd for C30H61N4O4 [M+H]+: 541.4687).
5,5′-Bis(1-dodecyl-4,10-bis(tert-butyloxycarbonyl)-1,4,7,10-tetraazacyclododecan-7-ylmethyl)-2,2′-bipyridine (18a)A mixture of 16a (0.27 g, 0.51 mmol), 5,5′-bis(bromomethyl)-2,2′-bipyridine 17 (0.086 g, 0.25 mmol) and sodium carbonate (0.32 g, 3.0 mmol) in MeCN (25 mL) was heated at reflux for 3 h under argon atmosphere. The reaction mixture was evaporated and the residue suspended in CHCl3 (50 mL). The organic layer was washed with H2O (30 mL) and brine, dried over Na2SO4, filtered and concentrated under reduced pressure. The remaining residue was purified by silica gel column chromatography (hexane–AcOEt=3 : 2) to give 18a as a pale yellow gum (0.23 g, 71% yield). IR (ATR) cm−1: 2925, 2854, 2801, 1687, 1460, 1410, 1365, 1247, 1151, 1047, 1027, 859, 753. 1H-NMR (300 MHz, CDCl3–TMS) δ: 8.53 (s, 2H), 8.27 (br d, 2H, J=7.8 Hz), 7.78 (d, 2H, J=7.2 Hz), 3.70 (s, 4H), 3.55–3.30 (m, 16H), 2.85–2.55 (m, 16H), 2.42–2.39 (m, 4H), 1.45 (m, 36H), 1.26 (m, 40H), 0.88 (t, 6H, J=6.9 Hz). 13C-NMR (75 MHz, CDCl3–TMS) δ: 155.9, 155.0, 149.5, 137.6, 134.9, 120.7, 79.4, 56.0, 55.4, 53.9, 47.2, 45.8, 31.9, 29.7, 29.7, 29.3, 28.4, 27.8, 26.5, 22.7, 14.1. Electrospray ionization (ESI)-MS m/z: 1261.9990 (Calcd for C72H129N10O8 [M+H]+: 1261.9989).
5,5′-Bis(1-dodecyl-1,4,7,10-tetraazacyclododecan-7-ylmethyl)-2,2′-bipyridine·8HBr·6H2O (19a)To a solution of 18a (0.22 g, 0.18 mmol) in MeOH (3 mL), 47% HBr (3 mL) was slowly added at 0°C. The mixture was stirred at room temperature for 12 h and then concentrated under reduced pressure. The remaining residue was precipitated from 47% HBr and MeOH to obtain 19a as a pale beige solid (221 mg, 78% yield). mp >250°C. IR (ATR) cm−1: 3381, 2923, 2853, 2583, 1632, 1600, 1573, 1551, 1443, 1066, 723. 1H-NMR (300 MHz, DMSO-d6–TMS) δ: 8.78 (s, 2H), 8.53 (d, 2H, J=8.1 Hz), 8.16 (d, 2H, J=8.1 Hz), 3.93 (s, 4H), 3.24 (br s, 8H), 3.13 (br s, 8H), 2.78 (m, 16H), 1.50–1.40 (m, 4H), 1.30–1.25 (m, 40H), 0.86 (t, 6H, J=6.9 Hz). 13C-NMR data could not be obtained because of the low solubility of this material. FAB-MS m/z: 861.7902 (Calcd for C52H97N10 [M+H]+: 861.7898) Anal. Calcd for C52H116Br8N10O3: C, 38.63; H, 7.23; N, 8.66. Found: C, 38.46; H, 7.16; N, 8.53.
6a·4NO3·5H2OAn aqueous mixture (5 mL) of 19a as HBr salt (0.10 g) was adjusted to pH >12 with a 2 N NaOH aqueous solution. The alkaline solution was extracted with CHCl3 (20 mL×3). The combined organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The resulting deprotonated 19a (48 mg, 56 µmol) was dissolved in EtOH (2 mL) and H2O (1.5 mL), to which a solution of Zn(NO3)2·6H2O (33 mg, 0.11 mmol) in EtOH was added. The solution was stirred at 70°C for 18 h. After evaporating the solvent, the residue was washed with AcOEt and EtOH to give 6a as a white solid (62 mg, 60% yield). mp >250°C. IR (ATR) cm−1: 3431, 3199, 2922, 2852, 1481, 1320, 1295, 1092, 1042, 1003, 959, 826. 1H-NMR (300 MHz, CD3OD–TMS) δ: 8.75–8.60 (m, 2H), 8.40–8.20 (m, 4H), 4.55–4.40 (m, 2H), 4.10–3.90 (m, 4H), 3.24–2.70 (m, 36H), 1.59 (m, 4H), 1.29 (m, 38H), 0.89 (t, 6H, J=6.9 Hz). 13C-NMR data could not be obtained because of the low solubility of this material. ESI-MS m/z: 1174.6026 (Calcd for C52H96N13O9Zn2 [M+(NO3)3]+: 1174.6031). Anal. Calcd for C52H106N14O17Zn2: C, 46.95; H, 8.03; N, 14.74. Found: C, 47.12; H, 7.67; N, 14.89.
1-Docosyl-4,10-bis(tert-butyloxycarbonyl)-1,4,7,10-tetraazacyclododecane (16b)A mixture of 1-bromodocosane 14b (0.75 g, 1.9 mmol) and sodium iodide (0.29 g, 1.9 mmol) in acetone (10 mL) and n-hexane (10 mL) was heated at reflux for 8 h under argon atmosphere. After the reaction, the insoluble compounds were filtered off and washed with n-hexanes (5 mL). The filtrate was concentrated under reduced pressure to give a white solid 15b (0.84 g), which was used in the next step without further purification. To a solution of 1,7-bis(tert-butyloxycarbonyl)-1,4,7,10-tetraazacyclododecane 13 (0.65 g, 1.7 mmol) and potassium carbonate (0.24 g, 1.7 mmol) in MeCN (10 mL) and CHCl3 (8 mL), 15b (0.84 g) in MeCN (2 mL) and CHCl3 (10 mL) was slowly added over a period of 15 min. The reaction mixture was stirred at room temperature for 3 d under argon atmosphere. After insoluble inorganic salts were filtered off, the filtrate was evaporated under reduced pressure. The remaining residue was purified by NH silica gel column chromatography (hexane–CHCl3=1 : 1) to give 16b as a colorless oil (0.52 g, 44% yield). IR (ATR) cm−1: 3225, 2923, 2853, 1694, 1459, 1407, 1365, 1246, 1160, 861, 754. 1H-NMR (300 MHz, CDCl3–TMS) δ: 3.40–3.25 (m, 8H), 2.85–2.62 (m, 8H), 2.53–2.47 (m, 2H), 1.45 (s, 18H), 1.25 (m, 40H), 0.88 (t, 3H, J=6.6 Hz). 13C-NMR (75 MHz, CDCl3–TMS) δ: 156.1, 155.8, 79.6, 79.4, 54.3, 53.7, 52.9, 50.3, 50.1, 49.8, 49.5, 49.2, 48.6, 48.4, 47.7, 47.4, 31.9, 29.8, 29.6, 29.6, 29.3, 28.4, 22.6, 14.1. FAB-MS m/z: 681.6256 (Calcd for C40H81N4O4 [M+H]+: 681.6258).
5,5′-Bis(1-docosyl-4,10-bis(tert-butyloxycarbonyl)-1,4,7,10-tetraazacyclododecan-7-ylmethyl)-2,2′-bipyridine (18b)A mixture of 16b (0.25 g, 0.37 mmol), 5,5′-bis(bromomethyl)-2,2′-bipyridine 17 (0.062 g, 0.18 mmol), and sodium carbonate (0.12 g, 1.1 mmol) in MeCN (10 mL) and CHCl3 (10 mL) was heated at reflux for 2 h under argon atmosphere. After the reaction mixture was evaporated, and suspended in CHCl3 (50 mL). The organic layer was washed with H2O (30 mL) and brine, dried over Na2SO4, filtered and evaporated under reduced pressure. The resulting residue was purified by silica gel column chromatography (hexane–AcOEt=3 : 2) to give 18b as a colorless oil (0.13 g, 46% yield). IR (ATR) cm−1: 2923, 2853, 1691, 1460, 1409, 1365, 1247, 1160, 1047, 1027, 754. 1H-NMR (300 MHz, CDCl3/TMS) δ: 8.53 (s, 2H), 8.27 (br d, 2H, J=7.8 Hz), 7.78 (d, 2H, J=7.8 Hz), 3.70 (s, 4H), 3.57–3.25 (m, 16H), 2.80–2.55 (m, 16H), 2.41 (t, 4H), 1.43 (m, 36H), 1.25 (m, 80H), 0.88 (t, 6H, J=6.6 Hz). 13C-NMR (75 MHz, CDCl3–TMS) δ: 155.8, 155.1, 149.5, 137.5, 134.8, 120.6, 79.3, 56.0, 55.4, 53.9, 53.7, 47.0, 45.7, 45.3, 31.9, 29.7, 29.3, 28.4, 27.7, 22.7, 14.1. ESI-MS m/z: 1542.3117 (Calcd for C92H169N10O8 [M+H]+: 1542.3119).
5,5′-Bis(1-docosyl-1,4,7,10-tetraazacyclododecan-7-ylmethyl)-2,2′-bipyridine·8HBr·3H2O (19b)To a solution of 18b (0.13 g, 86 µmol) in MeOH (4 mL) and CHCl3 (3 mL), 47% HBr (3 mL) was slowly added at 0°C. The resulting mixture was stirred at room temperature for 20 h, and then concentrated under reduced pressure. The remaining residue was recrystallized from 47% HBr and MeOH to give 19b as a pale beige solid (0.13 g, 80% yield). mp >250°C. IR (ATR) cm−1: 3412, 2885, 2850, 1553, 1471, 1395, 1145, 1085, 1047, 719. 1H-NMR (300 MHz, DMSO-d6–TMS) δ: 8.67 (s, 2H), 8.42 (d, 2H, J=8.1 Hz), 7.96 (d, 2H, J=8.1 Hz), 3.86 (s, 4H), 3.23–3.13 (m, 18H), 2.75 (br s, 18H), 1.46 (m, 4H), 1.23 (m, 80H), 0.85 (t, 6H, J=6.6 Hz). 13C-NMR data could not be obtained because of the low solubility of this material. ESI-MS m/z: 1142.1028 (Calcd for C72H137N10 [M+H]+: 1142.1022). Anal. Calcd for C72H150Br8N10O3: C, 46.92; H, 8.20; N, 7.60. Found: C, 46.70; H, 8.29; N, 7.66.
6b·4ClO4·2H2OAn aqueous mixture (5 mL) of 19b as HBr salt (40 mg) was adjusted to pH >12 with 2 N NaOH aqueous solution. The alkaline solution was extracted with CHCl3 (3×20 mL). The combined organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The resulting deprotonated 19b (26 mg, 23 µmol) was dissolved in EtOH (4 mL) and CHCl3 (3 mL), to which a solution of Zn(ClO4)2·6H2O (17 mg, 46 µmol) in H2O was added. The solution was stirred at 70°C for 36 h. After evaporating the solvent, the residue was crystallized from EtOH to give 6b as a white solid (37 mg, 99% yield). mp >250°C. IR (ATR) cm−1: 3527, 3277, 2919, 2851, 1467, 1086, 956, 926, 843, 748, 720, 621. 1H-NMR (300 MHz, CD3OD–TMS) δ: 8.68 (s, 2H), 8.42 (d, 2H, J=7.8 Hz), 7.99 (d, 2H, J=7.8 Hz), 4.10 (s, 4H), 3.07–2.80 (m, 32H), 1.62 (m, 4H), 1.29 (m, 80H), 0.90 (t, 6H, J=6.6 Hz) ppm. 13C-NMR data could not be obtained because of the low solubility of this material. Molecular-ion peaks corresponding to 6b were scarcely observed by ESI-MS. Anal. Calcd for C72H140Cl4N10O18Zn2: C, 50.68; H, 8.27; N, 8.21. Found: C, 50.30; H, 8.47; N, 8.07.
5,5-Bis(2-(4-(trifluoromethyl)benzyloxy)ethyl)barbituric Acid (25)To a solution of 2-((4-trifluoromethyl)benzyloxy)ethan-1-ol63) (0.76 g, 3.4 mmol) in distilled THF (12 mL) at 0°C, carbon tetrabromide (2.3 g, 6.9 mmol) was added and stirred for 5 min. After adding triphenylphosphine (1.8 g, 0.9 mmol), the reaction mixture was stirred for 1 h at room temperature, and then concentrated under reduced pressure. The resulting residue was purified by silica gel chromatography (hexane–AcOEt=50 : 1) to give 27 as colorless oil (0.95 g, 99%). IR (ATR) cm−1: 2864, 1622, 1421, 1322, 1162, 1105, 1064, 1017, 821. 1H-NMR (300 MHz, CDCl3–TMS) δ: 7.62 (d, 2H, J=8.2 Hz), 7.48 (d, 2H, J=8.2 Hz), 4.65 (s, 2H), 3.83 (t, 2H, J=6.0 Hz), 3.52 (t, 2H, J=6.0 Hz). 13C-NMR (75 MHz, CDCl3–TMS) δ: 142.0, 130.0, 127.6, 125.3, 72.9, 71.7, 61.6.
To a solution of diethyl malonate (0.26 g, 1.6 mmol) in distilled DMF (9 mL) at 0°C, NaH (0.12 mg, 4.8 mmol, 55 wt% dispersion in oil) was added and stirred for 5 min. After adding 27 (0.96 g, 3.4 mmol) in distilled DMF (3 mL), the reaction mixture was heated overnight at 70°C. The reaction was quenched with H2O, extracted with CHCl3 (30 mL×3), and the combined organic layers were washed with brine, dried over Na2SO4, and concentrated under reduced pressure. The resulting residue was purified by silica gel chromatography (hexane–AcOEt=30 : 1) to give 28 (0.32 g, 35%) as colorless oil. IR (ATR) cm−1: 2983, 2917, 1728, 1323, 1104, 1065, 822. 1H-NMR (300 MHz, CDCl3–TMS) δ: 7.58 (d, 4H, J=8.1 Hz), 7.40 (d, 4H, J=8.1 Hz), 4.47 (s, 4H), 4.08 (q, 6H, J=6.9 Hz), 3.54 (t, 4H, J=6.3 Hz), 2.33 (t, 4H, J=6.3 Hz), 1.17 (t, 4H, J=6.9 Hz). High resolution (HR)-MS (FAB) m/z: 565.2022 (Calcd for C27H31F6O6: 565.2019 [M+H]+).
To a slurry of NaH (22 mg, 0.91 mmol, 55 wt% dispersion in oil) and urea (96 mg, 1.6 mmol) in distilled DMF (2 mL) at 0°C, 28 (0.13 g, 0.22 mmol) in distilled DMF (3 mL) was added dropwise. The mixture was heated at 80°C for 12 h. After cooling to room temperature, the mixture was neutralized with cold saturated ammonium chloride solution and extracted with CHCl3. The reaction mixture was concentrated under reduced pressure, and the resulting solid was purified by silica gel chromatography (CHCl3) to afford 25 as white solid (64 mg, 53%). mp 158–159°C. IR (ATR) cm−1: 3276, 3101, 2870, 1704, 1319, 1113, 1122, 1063, 824, 497. 1H-NMR (300 MHz, CDCl3–TMS) δ: 7.59 (d, 4H, J=8.1 Hz), 7.51 (br s, 2H), 7.32 (d, 4H, J=8.1 Hz), 4.42 (s, 4H), 3.51 (t, 4H, J=5.7 Hz), 2.38 (t, 4H, J=5.7 Hz). 13C-NMR (75 MHz, CDCl3–TMS) δ: 171.9, 148.3, 141.3, 127.6, 125.4, 77.2, 72.5, 66.3, 51.6, 39.2. 19F-NMR (376 MHz, CDCl3–TFA) δ: −63.09. HR-MS (ESI) m/z: 533.1505 (Calcd for C24H23F6N2O5: 533.1506 [M+H]+). Anal. Calcd for C24H22F6N2O5: C, 54.14; H, 4.16; N, 5.26. Found: C, 54.23; H, 3.93; N, 5.26.
Extraction of MNP and BNP from the Aqueous Layer into a CHCl3 LayerA solution of a carrier (10b) (100 µM) in CHCl3 (1 mL) was prepared in situ. A solution of 7b (100 µM) in CHCl3 (1 mL) was added to 50 mM HEPES buffer (pH 7.4) with I=0.1 (NaNO3), and a solution of Cu(ClO4)2 (0.2 µmol) was then added and the resulting solution was mixed vigorously. For the extraction of MNP by 10b, 50 mM HEPES solution (pH 7.4, 4.0 mL) containing MNP or BNP (100 µM) was prepared and vigorously mixed with CHCl3 (1.0 mL) containing 10b (100 µM). The two layers were completely separated by centrifugation (3000 rpm×10 min at 25±0.1°C). The pH values of the aqueous layer remained unchanged before and after the extraction. The extraction efficiency of MNP or BNP was determined from UV absorption spectra (reproducibility ±1%). Control experiments without a carrier were conducted simultaneously.
Extraction of NP from an Aqueous Layer into a CHCl3 LayerFor the extraction of NP, 50 mM HEPES (pH 7.4, I=0.1 (NaNO3), 4.0 mL) containing 100 µM NP was prepared and vigorously mixed with CHCl3 (1 mL). The mixtures of two layers were completely separated by centrifugation (3000 rpm×10 min at 25±0.1°C). The pH values of the aqueous layer remained unchanged before and after the extraction. The extraction efficiency of NP was determined from UV/Vis absorption spectra (reproducibility±1%). These values were also checked by measuring the amount of transferred NP into CDCl3 (containing 100 µM naphthalene as an internal reference) by 1H-NMR. The partition ratio of NP is determined to be 79% in 50 mM HEPES buffer (pH 7.4, I=0.1 (NaNO3)).
Hydrolysis of MNP and BNPThe hydrolysis of MNP or BNP by supermolecules was carried out in n-butanol–50 mM HEPES buffer (pH 7.4) with I=0.1 (NaNO3) (8 : 2) (4, 5, 9a, 10a) and in CHCl3–50 mM HEPES buffer (pH 7.4) with I=0.1 (NaNO3) (8 : 2) (9b, 10b–h). Stock solutions of 1 (3.0 mM in H2O), CA (6.0 mM in H2O) or Bar (6.0 mM in n-butanol), and Cu(NO3)2·3H2O (9.4 mM in H2O) were used for preparation of sample solutions of 4 and 5 (final concentration: 20 µM or 100 µM in total solution) in n-butanol–50 mM HEPES buffer (pH 7.4) with I=0.1 (NaNO3) (8/2) (total volume 3.0 mL). Stock solutions of 6a (3.0 mM in n-butanol), CA (6.0 mM in H2O) or Bar (6.0 mM in n-butanol), and Cu(NO3)2·3H2O (9.4 mM in H2O) were used for preparation of sample solutions of 9a and 10a (final concentration: 20 or 100 µM in total solution) in n-butanol–50 mM HEPES buffer (pH 7.4) with I=0.1 (NaNO3) (8 : 2) (total volume 3.0 mL). Stock solutions of 6b (1.0 mM in CHCl3), CA (6.0 mM in H2O) or barbital derivative (Bar, 20–23, 25) (6.0 mM in CHCl3) or sodium salt of 24 (6.0 mM in H2O), and Cu(ClO4)2·6H2O (6.0 mM in H2O) were used for preparation of sample solutions of 9b and 10b–h (final concentration: 20 µM in total solution) in CHCl3–50 mM HEPES buffer (pH 7.4) with I=0.1 (NaNO3) (2 : 8) (total volume 3.0 mL). Stock aqueous solutions of MNP (20 or 100 mM) and BNP (100 mM) were used for hydrolysis reaction. All the hydrolysis experiments were performed at 37°C using shaking water bath (shaking speed: 196 rpm) (Tokyo Glass Kikai, FWB-1). In two-phase solvent system, the two layers of sample solutions were completely separated by centrifugation (3000 rpm×10 min at 25±0.1°C). The hydrolysis yields of MNP or BNP in the presence of the supermolecules were calculated based on the increase in the absorption of the released NP at 400 nm ([NP produced in the presence of the supermolecule]−[NP produced in the absence of the supermolecule]) (ε400 value of NP is 1.35×104 M−1·cm−1 at pH 7.4)70) in aqueous layer. The partition ratio of NP (79% in aqueous solution) was used for calculation of yields in CHCl3–H2O system.