Reviews
BET Bromodomain as a Target of Epigenetic Therapy
2016 年 64 巻 6 号 p. 540-547
詳細
2016 年 64 巻 6 号 p. 540-547
Acetylation of histone is a key epigenetic modification, and contributes to many DNA-dependent cellular processes. The bromodomain structure, which consists of approximately 110 amino acid residues, serves as a ‘reader’ that recognizes acetylated lysine in histones, leading to recruitment of positive transcriptional elongation factor b (P-TEFb), and thereby promoting transcriptional activity and chromatin remodeling. Among bromodomain-containing proteins, members of the bromodomain and extra-terminal domain (BET) family contain tandem N-terminal bromodomains. BET proteins, especially BRD4, have attracted interest as candidate therapeutic targets due to their putative involvement in the pathogenesis of various diseases, including cancer and inflammatory diseases. Several BET inhibitors are under clinical trial for treatment of various cancers. Furthermore, polypharmacological agents such as dual kinase/BET inhibitors and dual histone deacetylase (HDAC)/BET inhibitors have recently been developed, in addition to agents that degrade BET family proteins, such as proteolysis-targeting chimeras (PROTACs). This paper reviews recent progress in epigenetic therapy targeting the BET bromodomain.
Acetylation is a key histone modification that modulates many DNA-dependent cellular processes, including transcription, chromatin remodeling, the cell cycle, cell differentiation and DNA damage/repair. Histone acetylation is regulated by so-called ‘writers’ [such as histone acetyl transferase (HAT)], ‘readers’ (such as bromodomain-containing proteins), and ‘erasers’ [such as histone deacetylase (HDAC)].1) Dysfunction of these processes is considered to play a role in the pathogenesis of a diverse range of diseases, including cancer and inflammatory diseases. Therefore, small-molecular modulators of epigenetic regulators are expected to be candidates for epigenetic therapy. Indeed, several inhibitors of epigenetic ‘writers’ and ‘erasers’ have already been approved for clinical use by regulatory authorities such as the U.S. Food and Drug Administration (FDA) or are under clinical trial. However, development of inhibitors of epigenetic ‘readers’ has begun only in the last few years.2,3)
The bromodomain, which is a ‘reader’ of acetylated lysine in histones, was first identified as a result of studies on the Brahma gene in Drosophila,4,5) and it is now known that 61 bromodomains are encoded in the human genome. They are present in 46 diverse proteins, including histone acetyltransferases (HATs), chromatin-associated proteins, transcriptional coactivators, histone methyltransferases, and the bromodomain and extra-terminal domain (BET) family. The bromodomain is ca. 110-amino-acid module, which forms a conserved left-handed up-and-down four-helix bundle (helices αz, αa, αb and αc) with a pronounced cleft between two loops (termed the ZA loop and the BC loop)6,7) (Fig. 1A). The bromodomain recognizes Nε-acetylated lysine residue (Kac) of histones (Fig. 1B) and serves as a regulator of protein–protein interactions in numerous cellular processes, including transcription and chromatin remodeling.6,8,9) Therefore, the bromodomain has recently attracted attention as a candidate target for epigenetic therapy.

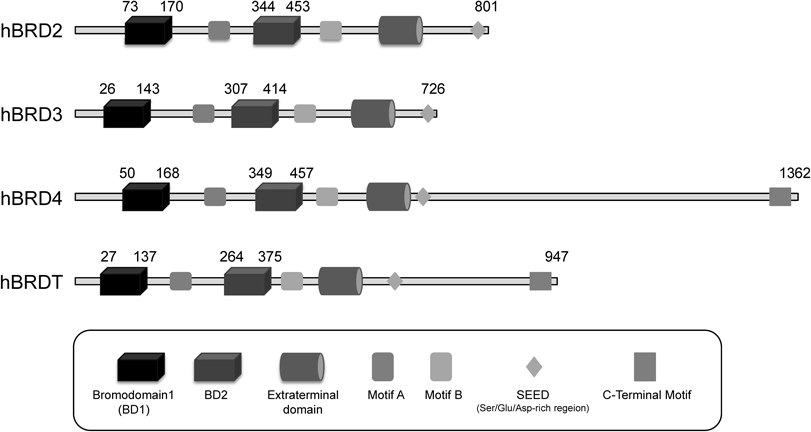
Most research aimed at developing small-molecular bromodomain inhibitors has so far been focused on the BET family proteins. The BET family consists of four members, BRD2, BRD3 and BRD4, which are expressed ubiquitously, and BRDT, whose expression is confined to the male germ cell. The functional differences among BRD2, BRD3, BRD4 and BRDT are still unclear. They each have tandem amino-terminal bromodomains9) (Fig. 2), and are normally localized in the cell nucleus; the conserved amino acid sequence between the two bromodomains of the BET family acts as a nuclear localization signal.10) The BET bromodomain interacts with Kac in histones such as H3 and H4.
BRD4 is the most extensively studied member of the BET family proteins. BRD4 binds Kac of histone via its bromodomains, and then recruits the positive transcription elongation factor b (P-TEFb), which is a heterodimer of cyclin-dependent kinase 9 (CDK9) and its regulator cyclin T, to the transcription start site, leading to transcription elongation through phosphorylation of the carboxyl-terminal of RNA polymerase II.11–13) BRD4 therefore acts as a transcriptional coactivator of many cellular genes. For example, the interaction of BRD4 with P-TEFb promotes G1 gene expression and cell cycle progression.14,15) BRD4 has also been reported to mark selected M/G1 genes for transcriptional memory during mitosis, enabling prompt postmitotic transcription in daughter cells.16) In addition, BRD4 modulates c-Myc transcriptional function, resulting in genome-wide regulation of Myc-dependent target genes.17) BRD4 has been suggested to influence differentiation induction via modulation of MYC in acute myelogenous leukemia (AML) cells, osteoclasts and osteoblasts.18,19) Therefore, BRD4 is considered to be a promising therapeutic target for treatment of AML and bone diseases.
BRD4 also binds to Kac of RelA, which is one of the subunits of nuclear factor-kappaB (NF-κB) transcriptional complex, thereby activating transcription of NF-κB and NF-κB-dependent inflammatory genes, including tumor necrosis factor (TNF)-α and interleukin (IL)-6, through the same mechanism as described above. Thus, BRD4 is also a candidate target for the treatment of inflammatory disorders.
Although the design of small molecules able to modulate protein–protein interactions is generally challenging,20,21) there have been many synthetic studies on BET bromodomain inhibitors since the first potent BET bromodomain inhibitors, (+)-JQ120) and I-BET762 (also known as GSK525762),21) were reported in 2010.22)
(+)-JQ1, a triazolothienodiazepine, is a BET-selective inhibitor and its chemical structure was inspired by and modified from similar BET inhibitors patented by Mitsubishi Tanabe Pharma.23) (+)-JQ1 is widely used in laboratory applications and various biological activities have been reported: (+)-JQ1 induces differentiation, G1 arrest and apoptosis in nuclear protein in testis (NUT) midline carcinoma (NMC) cells,20) and down-regulates MYC oncogene expression in multiple myeloma (MM) cells, consequently repressing MYC-dependent target genes.17) Potential applications of (+)-JQ1 in treatment of cancers, cardiovascular diseases, human immunodeficiency virus (HIV) infection, and inflammatory diseases have been proposed.
I-BET762, a triazolodiazepine, was developed by GlaxoSmithKline (GSK) at around the same time. It is also a BET-selective inhibitor, identified in an anti-inflammatory phenotypic screening search for enhancers of apolipoprotein A-1 (ApoA1) expression.21) I-BET762 reduces the expression of key inflammatory genes in lipopolysaccharide (LPS)-stimulated macrophages and suppresses inflammation in vivo.21) Moreover, I-BET762 down-regulates MYC expression in solid tumors, including prostate cancer cell lines and myeloma cells that frequently overexpress MYC, and up-regulates HEXIM1, a negative regulator of P-TEFb, leading to induction of cell cycle arrest and apoptosis of myeloma cells.24,25) I-BET762 is in phase 1 clinical trial for treatment of NMC and other cancers (Fig. 3).
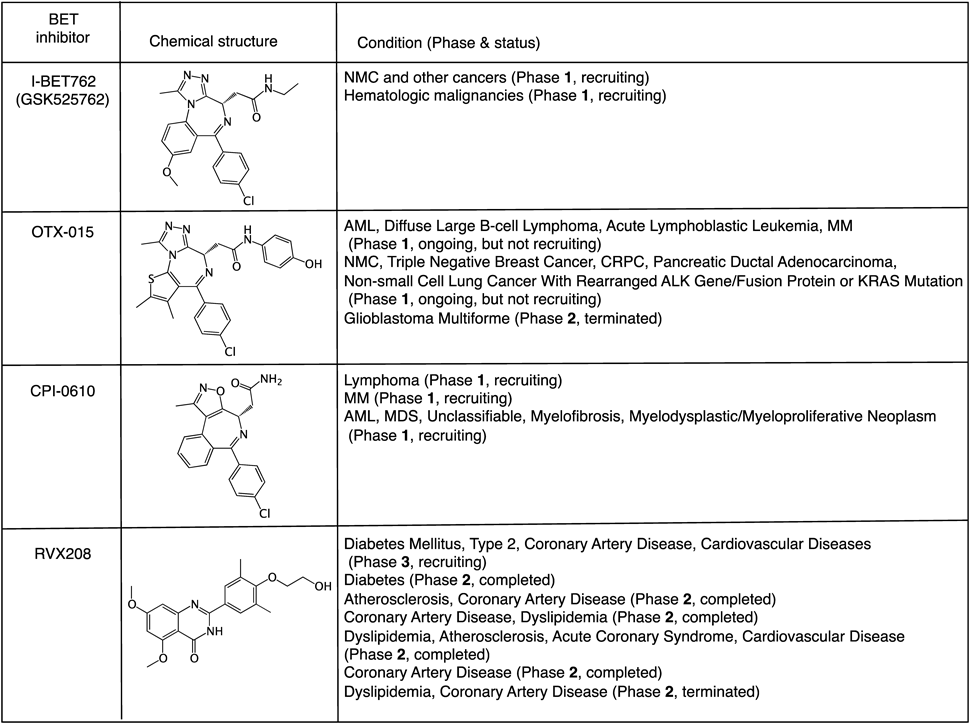
Several other BET inhibitors are also in clinical trials, as summarized in Fig. 3. For example, RVX-208 (also known as RVX000222), a quinazolone, is under phase 3 clinical trial in high-risk type 2 diabetes mellitus patients with coronary artery diseases and cardiovascular diseases. Interestingly, RVX-208 appears to bind selectively to the second bromodomain (BD2) of BRD4. OTX-015, a triazolothienodiazepine like (+)-JQ1, is in phase 1B clinical trial for treatment of selected advanced solid tumors, including NMC and castration-resistant prostate cancer (CRPC). CPI-0610 is in Phase 1 clinical trial for treatment of lymphoma, MM, myelodysplastic syndrome (MDS), and myelofibrosis.
3.2. Kac Binding Mode with the BET BromodomainThe tyrosine (Tyr) residue in the ZA loop and the asparagine (Asn) residue in the BC loop are involved in Kac recognition, and are conserved in diverse bromodomains.6,8,9,22) Kac forms a hydrogen bond with the Asn residue and a water-mediated hydrogen bond with the Tyr residue. The hydrophobic WPF shelf, consisting of tryptophan (Trp, W), proline (Pro, P) and phenylalanine (Phe, F) residues in the ZA loop, and the gatekeeper residue located at the entrance to the Kac direct binding site are also well-conserved, and are important for Kac binding and selectivity.22)
Known BET inhibitors bind to the Kac-binding site of BET bromodomain. As can be seen in the crystal structures (Fig. 4), both (+)-JQ1 and I-BET762 bind directly to the conserved Asn (N140 in BRD4) and form a water-mediated hydrogen bond with the conserved Tyr (Y97 in BRD4) via the triazole moiety. The chlorophenyl substituent of (+)-JQ1 fits into a shelf formed by W81, P82 and D145, while its dimethyl-substituted thieno moiety fits into a shelf formed by W81, P82 and L92, stabilizing the interaction. The selectivity for BET bromodomain depends on the presence of the hydrophobic pocket in the ZA loop, including the WPF shelf (W81, P82, F83 in BRD4), and the gatekeeper residue (I146 in BRD4).20,21)

These crystallographic data have been utilized to develop other types of BET inhibitors with Kac-mimetic structures, including derivatives of dimethylisoxazole, quinazoline,26) pyridone27) and purine28,29) (Fig. 5). For example, we developed purine derivatives based on thalidomide structure and examined the structure–activity relationship of methoxy-substituted derivatives29) (Fig. 5). Nodes in the figure represent substitution sites or site combinations that are scaled in size/color density according to the potency of BRD4-inhibitory activity. N6-(2,4,5-Trimethoxybenzoyl) adenine was the most potent inhibitor among our N6-benzoyladenine derivatives, being 50-fold more potent than N6-benzoyladenine.

Various nuclear receptors are involved in mediating transcriptional regulation by BET bromodomain.
In castration-resistant prostate cancer (CRPC) cells, BRD4 binds directly to the amino-terminal domain of androgen receptor (AR). The BET inhibitor (+)-JQ1 disrupts the interaction between BRD4 and AR, blocking AR recruitment to target gene loci and thus decreasing AR-mediated gene transcription. Furthermore, (+)-JQ1 significantly reduce tumor volume and weight in a mouse model of vertebral cancer of the prostate (VCaP).30)
BRD4 also regulates estrogen receptor (ER)-dependent gene transcription and the recruitment and elongation of RNA polymerase II on estrogen response elements.31) The BET inhibitor JQ1 suppressed estradiol-induced growth and transcription in ER-positive breast cancer cell lines by blocking transition of RNA polymerase II at the promoter region upstream of the transcription start site.32) BRD4 also serves as a transcriptional coactivator of nuclear retinoic acid receptors (RARs) and functions in retinoid-mediated differentiation of neuroblastoma.33,34)
On the other hand, BRD2 binds to complexes of peroxisome proliferator-activated receptor (PPAR)-γ in a ligand-independent manner and co-represses PPAR-γ. It may play roles in diabetes and obesity.35)
Polypharmacology has recently attracted interest as a robust therapeutic strategy in which a single drug molecule simultaneously modulates multiple disease-associated targets. The polypharmacological approach might be particularly suitable for the treatment of diseases with complex pathogenic mechanisms, including cancers and inflammatory diseases. Advantages could include reduced cost and reduced side effects as compared to classical combination therapy. Polypharmacological agents for which the bromodomain is one target are discussed below.
4.1. Dual Kinase/BET InhibitorsThe BET bromodomain functions as an atypical kinase that phosphorylates serine in the carboxyl-terminal domain of RNA polymerase II, and it contains regions with weak similarity to other kinase motifs.36) Indeed, several kinase inhibitors with potent bromodomain–inhibitory activity were identified by screening a collection of diverse kinase inhibitors against BRD4(1).37) They include clinically advanced compounds such as BI 2536 and BI 6727 (also known as volasertib), which are ATP-competitive polo-like kinase (PLK) inhibitors, TG-101348 (also known as fedratinib), which is an ATP-competitive Janus kinase (JAK) inhibitor, and Tideglusib, which is a non-ATP-competitive glycogen synthase kinase (GSK)-3 inhibitor. Several co-crystal structures of BRD4(1) complexes with kinase inhibitors showing potent bromodomain-inhibitory activity have been reported.37–39)
Both BI 2536 and LY294002, which is a ATP-competitive phosphoinositide 3-kinase (PI3K) inhibitor, are anchored to the hinge region of the ATP-binding site in kinase by a hydrogen bond.37,39,40) In addition, both the dihydropteridinone carbonyl oxygen in Kac-mimetic BI 2536 and the morpholino oxygen in LY294002 form hydrogen bonds with the conserved Asn (N140) in the Kac-binding pocket of BRD4; the co-crystal structures of BI 2536/LY294002 and BRD4(1) are illustrated in Fig. 6. BI 6727, which is a derivative of BI 2536 and a dual PLK kinase/BET inhibitor, is under clinical trial for treatment of leukemia and MDS. The results of this study will have important implications for the future of polypharmacological dual kinase/BET inhibitors.

Diverse synergistic drug interactions of BET inhibitors have been reported, e.g., with kinase inhibitors, histone methyltransferases, and DNA intercalators. Also, many studies have shown that HDAC inhibitors have a synergistic anti-tumor effect with BET inhibitors. For instance, the combination of Panobistat or Vorinostat (also known as SAHA), clinically used HDAC inhibitors, with (+)-JQ1, a BET inhibitor, enhances apoptosis of AML and pancreatic ductal adenocarcinoma (PDAC) cells. Recently, potent dual HDAC/BET inhibitors have been developed. One of them, DUAL946, contains a hydroxamic acid group for binding with HDAC and a 1-formyl-1,2,3,4-tetrahydroquinoline moiety for binding with BET (Fig. 7). It inhibits LPS-induced TNF-alpha and IL-6 production and down-regulates c-myc expression.41)

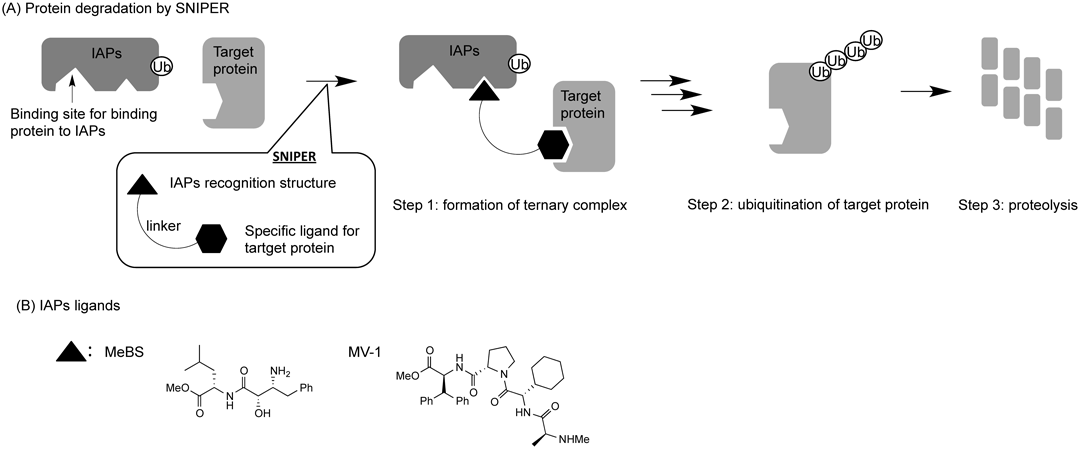
In contrast to conventional approaches that regulate functions elicited by the target protein, an alternative approach is to utilize spatiotemporal regulators of target protein synthesis or degradation. For example, posttranslational degradation of target protein can be achieved through the recruitment of E3 ubiquitin ligases with PROTACs, which bear a peptide E3 ligase–recognition structure.42–46) Another strategy is called SNIPER (specific and non-genetic inhibitor of apoptosis protein (IAP)-dependent protein eraser)47–51); SNIPERs are bifunctional compounds in which a small IAP-recognition structure, such as MeBS or MV-1, is conjugated with a target protein–recognition structure47–51) (Fig. 8).
MZ1, dBET1 and ARV-825 have been developed as BET family protein degradation modulators (Fig. 9). dBET1 and ARV-825 consist of BET inhibitors, (+)-JQ1 and OTX-015, respectively, coupled via a spacer moiety to a ligand of E3 ubiquitin ligase.52–55) dBET1 induced selective CUL4-RBX1-DDB1-cereblon (CRBN)-dependent degradation of BET family proteins, including BRD2, BRD3 and BRD4, in vitro and in vivo, and it delayed leukemia progression in mice.54) ARV-825 induced BRD4 degradation via a CRBN-mediated and proteasome-dependent mechanism, leading to persistent suppression of c-MYC and its downstream signaling.55) ARV-825 inhibited cell proliferation and induced apoptosis in Burkitt’s lymphoma (BL), which is MYC-driven malignancy.55) Both dBET1 and ARV-825 were more effective against cancer cells than JQ1 or OTX-015 alone. Degradation of BET family proteins by MZ1, which utilizes a different E3 ligase system, has also been reported. MZ1 is a hybrid compound consisting of VHL-1, a ligand of E3 ligase von Hipple-Lindau protein (VHL), and the BET inhibitor (+)-JQ1.56) Interestingly, MZ1 induces selective degradation of BRD4 over BRD2 and BRD3, although (+)-JQ1 is a non-selective inhibitor toward bromodomains of BET family proteins.

The BET bromodomain, which functions as a ‘reader’ of acetylated lysine in histones, is a promising target for epigenetic therapy to modulate transcriptional activity and chromatin remodeling. This review has focused on the current status of BET bromodomain inhibitors. Several BET inhibitors are already under clinical trial for treatment of various cancers and other diseases. Recently, there has been increasing focus on polypharmacology, and a number of dual kinase/BET inhibitors and dual HDAC/BET inhibitors have been developed. In addition, PROTACs targeting BET proteins for degradation have been reported. We expect continuing rapid development of both monospecific BET inhibitors and multi-target BET inhibitors as candidate therapeutic agents for cancers, inflammatory diseases, immune deficiency diseases, diabetes, and cardiovascular diseases. Highly specific inhibitors of individual BET proteins would also be useful tools for investigating the functions of BRD2, BRD3 and BRD4 and the roles of the tandem bromodomains, as well as being candidate therapeutic agents.
The work described in this paper was partially supported by Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan, and a Grant from the Japan Society for the Promotion of Science.
The author declares no conflict of interest.