Experimental
All melting points were determined on a Yanagimoto melting point apparatus and are uncorrected. Optical rotations were recorded on a JASCO P-2300 polarimeter. IR spectra were recorded on a Nicolet AVATAR 320 FT-IR spectrometer. 1H- and 13C-NMR spectra were recorded on a Bruker AV-400M (400 MHz) or DRX-500 (500 MHz) spectrometer using tetramethylsilane as the internal standard. High-resolution (HR)-MS were recorded on an Agilent Technologies Q-TOF 6520 mass spectrometer.
General Procedure for the Preparation of 4-(Substituted benzyl)-1,2-dihydro-5-methyl-3H-pyrazol-3-ones (3a–c)N,N-Diisopropylethylamine (12.9 g, 100 mmol) was added dropwise to a mixture of an appropriate benzyl chloride 6a–c (50 mmol), methyl acetoacetate (7) (6.39 g, 55 mmol), lithium chloride (2.33 g, 55 mmol), and potassium iodide (0.896 g, 5.4 mmol) in a mixed solvent of toluene (20 mL) and DMF (20 mL) at 50°C with stirring. The reaction mixture was stirred at 70°C for 12 h and then cooled to room temperature. After the addition of toluene (20 mL), the resulting mixture was washed successively with water (30 mL), 2 M HCl (30 mL), water (30 mL), saturated aqueous NaHCO3 (30 mL), and water (30 mL). The organic layer was concentrated under reduced pressure. Hydrazine monohydrate (3.25 g, 65 mmol) was added dropwise to the residual solution in toluene (30 mL) at room temperature. The reaction mixture was stirred for 12 h at 70°C and then cooled to room temperature. n-Hexane (30 mL) and 2-propanol (5 mL) were then added to the mixture in that order. The resulting slurry was stirred for 1 h at room temperature and for an additional 1 h at 0°C. The precipitate was filtered off and dried in vacuo to provide 3a–c.
1,2-Dihydro-4-[(4-isopropoxyphenyl)methyl]-5-methyl-3H-pyrazol-3-one (3a)White solid (71% from 6a). mp 227–234°C (decomp.). IR (KBr) cm−1: 2977, 1609, 1543, 1507, 1477, 1420, 1383, 1372. 1H-NMR (DMSO-d6) δ: 1.22 (6H, d, J=6.0 Hz), 2.00 (3H, s), 3.46 (3H, s), 4.46–4.55 (1H, m), 6.77 (2H, d, J=8.8 Hz), 7.04 (2H, d, J=8.8 Hz), 10.02–10.46 (2H, br s). 13C-NMR (DMSO-d6) δ: 9.86 (q), 21.77 (q×2), 26.28 (t), 68.87 (d), 100.30 (s), 115.26 (d×2), 128.83 (d×2), 133.60 (s), 136.65 (s), 155.22 (s), 159.55 (s). HR-MS (electrospray ionization (ESI)) m/z: 247.1448 [M+H]+ (Calcd for C14H19N2O2: 247.1441).
1,2-Dihydro-4-[(3-fluoro-4-methoxyphenyl)methyl]-5-methyl-3H-pyrazol-3-one (3b)White solid (69% from 6b). mp 216–220°C (decomp.). IR (KBr) cm−1: 1609, 1518, 1435, 1275. 1H-NMR (DMSO-d6) δ: 2.01 (3H, s), 3.49 (2H, s), 3.78 (3H, s), 6.90–6.97 (2H, m), 7.02 (1H, t, J=8.7 Hz), 10.39 (2H, br s). 13C-NMR (DMSO-d6) δ: 9.79 (q), 26.17 (t), 55.85 (q), 99.74 (s), 113.55 (d, JCF=1.5 Hz), 115.22 (d, JCF=17.6 Hz), 123.68 (d, JCF=2.9 Hz), 134.98 (d, JCF=5.8 Hz), 136.83 (s), 144.83 (d, JCF=10.3 Hz), 151.17 (d, JCF=244.3 Hz), 159.55 (s). HR-MS (ESI) m/z: 237.1030 [M+H]+ (Calcd for C12H14FN2O2: 237.1034).
1,2-Dihydro-4-[(2-fluoro-4-methoxyphenyl)methyl]-5-methyl-3H-pyrazol-3-one (3c)White solid (64% from 6c). mp 226–227°C (decomp.). IR (KBr) cm−1: 1628, 1598, 1512, 1466, 1446, 1435, 1409. 1H-NMR (DMSO-d6) δ: 2.00 (3H, s), 3.48 (2H, s), 3.71 (3H, s), 6.67 (1H, dd, J=2.4, 8.4 Hz), 6.74 (1H, dd, J=2.9, 12 Hz), 7.02 (1H, t, J=8.8 Hz), 10.38 (2H, br s). 13C-NMR (DMSO-d6) δ: 9.71 (q), 19.41 (d, JCF=2.9 Hz), 55.34 (q), 98.48 (s), 101.00 (d, JCF=25.7 Hz), 109.76 (d, JCF=2.9 Hz), 119.70 (d, JCF=16.1 Hz), 130.58 (d, JCF=7.4 Hz), 136.85 (s), 158.53 (d, JCF=11.0 Hz), 159.68 (s), 160.34 (d, JCF=242.9 Hz). HR-MS (ESI) m/z: 237.1031 [M+H]+ (Calcd for C12H14FN2O2: 237.1034).
1-Acetyl-1,2-dihydro-4-[(4-isopropoxyphenyl)methyl]-5-methyl-3H-pyrazol-3-one (10a)Acetic anhydride (3.27 g, 32 mmol) was added dropwise to a mixture of 3a (7.39 g, 30 mmol) and K2CO3 (4.56 g, 33 mmol) in DMF (27 mL) at 40°C. The reaction mixture was stirred for 30 min at 40°C and for 1 h at 70°C. The reaction mixture was cooled to 50°C and the inorganic salt was filtered off and washed with DMF (12 mL). Acetic acid (0.180 g, 3.0 mmol) in H2O (1.8 mL) was added to the combined filtrates and stirred for 30 min at 20°C. H2O (30 mL) was added to the resulting slurry and the mixture was stirred for 1 h at 20°C. The precipitate was filtered off and dried in vacuo at 60°C to provide 10a (7.01 g, 81%) as a white solid. mp 166–173°C (decomp.). IR (KBr) cm−1: 2978, 2924, 1725, 1626, 1609, 1539, 1507, 1396, 1377, 1318, 1307. 1H-NMR (DMSO-d6) δ: 1.22 (6H, d, J=6.0 Hz), 2.41 (3H, s), 2.45 (3H, s), 3.52 (2H, s), 4.47–4.56 (1H, m), 6.79 (2H, d, J=8.7 Hz), 7.05 (2H, d, J=8.7 Hz), 10.97 (1H, br s). 13C-NMR (DMSO-d6) δ: 12.84 (q), 21.77 (q×2), 23.16 (q), 25.75 (t), 68.93 (d), 111.25 (s), 115.46 (d×2), 128.93 (d×2), 131.70 (s), 139.97 (s), 155.62 (s), 161.62 (s), 169.88 (s). HR-MS (ESI) m/z: 289.1541 [M+H]+ (Calcd for C16H21N2O3: 289.1547).
1-Acetyl-3-benzyloxy-4-[(4-isopropoxyphenyl)methyl]-5-methyl-1H-pyrazole (12)A mixture of 10a (4.32 g, 15 mmol), benzyl bromide (3.08 g, 18 mmol), and K2CO3 (3.11 g, 22.5 mmol) in MeCN (20 mL) was stirred at 60°C for 2 h and the resulting inorganic salt was filtered off. The filtrate was concentrated under reduced pressure and the residue was purified by silica gel chromatography (eluent AcOEt–n-hexane, 1 : 3) to provide 12 (3.89 g, 69%) as a colorless oil. IR (KBr) cm−1: 2975, 2934, 1718, 1612, 1507, 1467, 1453, 1375, 1337. 1H-NMR (CDCl3) δ: 1.31 (6H, d, J=6.0 Hz), 2.84 (3H, s), 2.56 (3H, s), 3.56 (2H, s), 4.44–4.53 (1H, m), 5.26 (2H, s), 6.77 (2H, d, J=8.5 Hz), 7.06 (2H, d, J=8.5 Hz), 7.30–7.35 (5H, m). 13C-NMR (CDCl3) δ: 13.14 (q), 22.07 (q×2), 23.37 (q), 26.50 (t), 69.86 (d), 70.07 (t), 111.38 (s), 115.85 (d×2), 127.84 (d×2), 127.93 (d), 128.34 (d×2), 129.17 (d×2), 131.69 (s), 136.83 (s), 141.24 (s), 156.24 (s), 162.54 (s), 170.98 (s). HR-MS (ESI) m/z: 379.2018 [M+H]+ (Calcd for C23H27N2O3: 379.2016).
3-Benzyloxy-4-[(4-isopropoxyphenyl)methyl]-1,5-dimethyl-1H-pyrazole (14)A mixture of 12 (3.60 g, 9.51 mmol) and KHCO3 (0.286 g, 2.85 mmol) in MeOH (20 mL) was stirred at 50°C for 2 h and the reaction mixture was concentrated under reduced pressure. AcOEt (40 mL) was added to the residue and the mixture was washed with H2O (20 mL). The organic layer was dried over MgSO4 and the filtrate was concentrated under reduced pressure to provide an oil. The oil was dissolved in DMAc (10 mL) and added dropwise to a suspension of sodium hydride (0.571 g, 14.3 mmol, 60% oil dispersion) in DMAc (5 mL) at 0°C under a N2 atmosphere and the mixture was stirred for 15 min at 0°C. MeI (2.70 g, 19.0 mmol) was added and the mixture was stirred for 2 h. The reaction mixture was diluted with AcOEt (70 mL) and washed with H2O (30 mL×2). The resulting organic layer was dried over MgSO4 and the filtrate was concentrated under reduced pressure. The residue was purified by silica gel chromatography (eluent AcOEt–n-hexane, 1 : 2) to provide 14 (2.84 g, 85% yield) as a colorless oil. IR (KBr) cm−1: 2974, 2934, 1738, 1611, 1580, 1506, 1464, 1453, 1383, 1365, 1333. 1H-NMR (CDCl3) δ: 1.30 (2H, d, J=6.0 Hz), 2.07 (3H, s), 3.60 (2H, s), 3.62 (3H, s), 4.43–4.52 (1H, m), 5.21 (2H, s), 6.74 (2H, d, J=8.6 Hz), 7.08 (2H, d, J=8.6 Hz), 7.26–7.39 (5H, m). 13C-NMR (CDCl3) δ: 9.88 (q), 22.10 (q×2), 27.21 (t), 35.56 (q), 69.86 (d), 69.97 (t), 102.40 (s), 115.76 (d×2), 127.45 (d×2), 127.55 (d), 128.28 (d×2), 129.10 (d×2), 133.52 (s), 137.45 (s), 137.77 (s), 155.93 (s), 160.17 (s). HR-MS (ESI) m/z: 351.2071 [M+H]+ (Calcd for C22H27N2O2: 351.2067).
1,2-Dihydro-4-[(4-isopropoxyphenyl)methyl]-1,5-dimethyl-3H-pyrazol-3-one (10b)A solution of 14 (2.70 g, 7.70 mmol) in THF (30 mL) was hydrogenated over 10% Pd/C (50% wet, 0.30 g) for 10 h at room temperature under atmospheric pressure. The Pd–C was filtered off and the filtrate was concentrated under reduced pressure to provide 10b (1.98 g, 99%) as a white solid. mp 160–162°C. IR (KBr) cm−1: 1613, 1540, 1511, 1372, 1300. 1H-NMR (DMSO-d6) δ: 1.22 (6H, d, J=6.0 Hz), 2.03 (3H, s), 3.45 (2H, s), 3.47 (3H, s), 4.46–4.55 (1H, m), 6.76 (2H, d, J=8.5 Hz), 7.03 (2H, d, J=8.5 Hz), 9.37 (1H, br s). 13C-NMR (DMSO-d6) δ: 9.31(q), 21.76 (q×2), 26.56 (t), 34.97 (q), 68.87 (d), 100.95 (s), 115.27 (d×2), 128.76 (d×2), 133.59 (s), 136.22 (s), 155.24 (s), 157.91 (s). HR-MS (ESI) m/z: 261.1609 [M+H]+ (Calcd for C15H21N2O2: 261.1598).
1-Acetyl-4-[(4-isopropoxyphenyl)methyl]-5-methyl-3-(2,3,4,6-tetra-O-acetyl-β-D-glucopyranosyloxy)-1H-pyrazole (11a)5a (0.987 g, 2.4 mmol) was added to a mixture of 10a (0.577 g, 2.0 mmol) and K2CO3 (0.387 g, 2.8 mmol) in MeCN (5 mL), and the reaction mixture was stirred for 16 h at 60°C. The mixture was diluted with AcOEt (20 mL) and washed with H2O (5 mL). The resulting organic layer was concentrated under reduced pressure. The residue was purified by silica gel chromatography (eluent AcOEt–n-hexane, 1 : 3) to provide 11a (1.11 g, 90% yield). An analytical sample of 11a was obtained as a white solid by recrystallization from Et2O. mp 68–72°C. [α]D20 −19.3 (c=1.0, dimethyl sulfoxide (DMSO)). IR (KBr) cm−1: 3436, 2977, 2934, 1759, 1611, 1509, 1472, 1431, 1373, 1336. 1H-NMR (CDCl3) δ: 1.29 (3H, dd, J=3.8, 6.2 Hz), 1.86 (3H, s), 2.02 (3H, s), 2.04 (3H, s), 2.06 (3H, s), 2.50 (3H, s), 2.55 (3H, s), 3.54 (2H, dd, J=15.6, 19.9 Hz), 3.86–3.91 (1H, m), 4.16 (1H, dd, J=2.3, 12.5 Hz), 4.27 (1H, dd, J=4.8, 12.2 Hz), 4.43–4.52 (1H, m), 5.18 (1H, t, J=9.6 Hz), 5.24–5.33 (2H, m), 5.71 (1H, d, J=7.8 Hz), 6.75 (2H, d, J=8.5 Hz), 7.01 (2H, d, J=8.5 Hz). 13C-NMR (CDCl3) δ: 13.12 (q), 20.41 (q), 20.60 (q×2), 20.71 (q), 22.03 (q), 22.09 (q), 23.19 (q), 26.25 (t), 61.82 (t), 68.17 (d), 69.87 (d), 70.62 (d), 72.48 (d), 72.83 (d), 96.67 (d), 111.39 (s), 115.93 (d×2), 129.13 (d×2), 131.30 (s), 141.76 (s), 156.31 (s), 160.45 (s), 169.15 (s), 169.42 (s), 170.23 (s), 170.61 (s), 170.85 (s). HR-MS (ESI) m/z: 619.2473 [M+H]+ (Calcd for C30H39N2O12: 619.2498).
4-[(4-Isopropoxyphenyl)methyl]-1,5-dimethyl-3-(2,3,4,6-tetra-O-acetyl-β-D-glucopyranosyloxy)-1H-pyrazole (11b)5a (0.987 g, 2.4 mmol) was added to a mixture of 10b (0.521 g, 2.0 mmol) and K2CO3 (0.387 g, 2.8 mmol) in MeCN (5 mL) and the reaction mixture was stirred for 16 h at 60°C. The reaction mixture was diluted with AcOEt (20 mL) and washed with H2O (5 mL). The resulting organic layer was concentrated under reduced pressure. The residue was purified by silica gel chromatography (eluent AcOEt–n-hexane, 1 : 1) to provide 11b (0.311 g, 26%). An analytical sample of 11b was obtained as a white solid by recrystallization from Et2O-n-hexane. mp 65–69°C. [α]D20 −11.2 (c=1.0, DMSO). IR (KBr) cm−1: 2977, 1759, 1751, 1508, 1496, 1371. 1H-NMR (CDCl3) δ: 1.29 (6H, dd, J=1.9, 6.3 Hz), 1.90 (3H, s), 2.01 (3H, s), 2.03 (3H, s), 2.06 (3H, s), 2.07 (3H, s), 3.54 (2H, dd, J=16.0, 14.7 Hz), 3.60 (3H, s), 3.82–3.86 (1H, m), 4.09–4.16 (1H, m), 4.30 (1H, dd, J=4.3, 12.5 Hz), 4.42–4.51 (1H, m), 5.17–5.29 (3H, m), 5.53–5.55 (1H, m), 6.74 (2H, d, J=8.7 Hz), 7.02 (2H, d, J=8.7 Hz). 13C-NMR (CDCl3) δ: 9.92 (q), 20.53 (q), 20.62 (q), 20.65 (q), 20.75 (q), 22.07 (q), 22.10 (q), 26.88 (t), 35.77 (q), 61.71 (t), 68.18 (d), 69.85 (d), 70.98 (d), 71.94 (d), 72.96 (d), 97.76 (d), 103.62 (s), 115.78 (d×2), 129.06 (d×2), 133.09 (s), 137.63 (s), 155.97 (s), 157.67 (s), 169.34 (s), 169.45 (s), 170.27 (s), 170.77 (s). HR-MS (ESI) m/z: 591.2554 [M+H]+ (Calcd for C29H39N2O11: 591.2548).
4-[(4-Isopropoxyphenyl)methyl]-5-methyl-3-(2,3,4,6-tetra-O-acetyl-β-D-glucopyranosyloxy)-1H-pyrazole (2a)A mixture of 11a (0.618 g, 1.0 mmol) and KHCO3 (0.030 g, 0.30 mmol) in MeOH (3 mL) was stirred at 50°C for 1 h; AcOH (0.018 g, 0.30 mmol) was then added at room temperature. After the reaction mixture was concentrated under reduced pressure, the residue was purified by silica gel chromatography (eluent AcOEt–n-hexane, 1 : 2) to provide 2a (0.259 g, 45% yield). An analytical sample of 2a was obtained as a white solid by recrystallization from Et2O. mp 155–156°C. [α]D20 −7.9 (c=1.0, DMSO). IR (KBr) cm−1: 3393, 3222, 2978, 2943, 1751, 1735, 1612, 1509, 1474, 1434, 1374. 1H-NMR (CDCl3) δ: 1.30 (6H, dd, J=2.3, 5.9 Hz), 1.89 (3H, s), 2.01 (3H, s), 2.03 (3H, s), 2.05 (3H, s), 2.10 (3H, s), 3.55 (2H, dd, J=15.7, 11.7 Hz), 3.83–3.87 (1H, m), 4.11 (1H, dd, J=2.3, 12.5 Hz), 4.31 (1H, dd, J=4.1, 12.3 Hz), 4.43–4.52 (1H, m), 5.18–5.30 (3H, m), 5.58 (1H, d, J=6.8 Hz), 6.75 (2H, d, J=8.8 Hz), 7.02 (2H, d, J=8.8 Hz), 9.09 (1H, br s). 13C-NMR (CDCl3) δ: 10.36 (q), 20.48 (q), 20.61 (q), 20.63 (q), 20.72 (q), 22.07 (q), 22.10 (q), 26.50 (t), 61.64 (t), 68.07 (d), 69.89 (d), 70.93 (d), 72.06 (d), 72.92 (d), 97.67 (d), 103.54 (s), 115.84 (d×2), 129.12 (d×2), 132.69 (s), 138.21 (s), 156.05 (s), 159.89 (s), 169.31 (s), 169.44 (s), 170.30 (s), 170.76 (s). HR-MS (ESI) m/z: 577.2380 [M+H]+ (Calcd for C28H37N2O11: 577.2392).
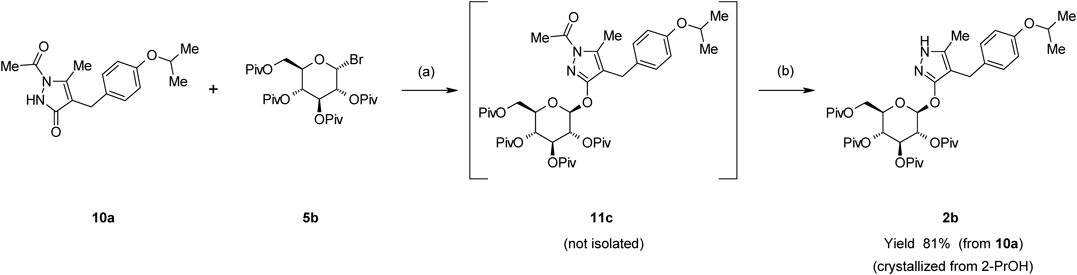
1-Acetyl-4-[(4-isopropoxyphenyl)methyl]-5-methyl-3-(2,3,4,6-tetra-O-pivaloyl-β-D-glucopyranosyloxy)-1H-pyrazole (11c)5b (1.39 g, 2.4 mmol) was added to a mixture of 10a (0.577 g, 2.0 mmol) and K2CO3 (0.387 g, 2.8 mmol) in MeCN (5 mL), and the mixture was stirred for 16 h at 60°C. The reaction mixture was diluted with AcOEt (20 mL) and washed with H2O (5 mL). The resulting organic layer was concentrated under reduced pressure. The obtained residue was purified by silica gel chromatography (eluent AcOEt–n-hexane, 1 : 3) to provide 11c (1.44 g, 92% yield). An analytical sample of 10c was obtained as a white solid by recrystallization from n-hexane. mp 137–140°C. [α]D20 −2.0 (c=1.0, DMSO). IR (KBr) cm−1: 3469, 2975, 2936, 2875, 1747, 1612, 1509, 1480, 1470, 1398, 1370, 1333. 1H-NMR (CDCl3) δ: 1.01 (9H, s), 1.13 (9H, s), 1.16 (9H, s), 1.18 (9H, s), 1.29 (6H, dd, J=2.4, 6.1 Hz), 2.47 (3H, s), 2.54 (3H, s), 3.53 (2H, s), 3.88–3.92 (1H, m), 4.12–4.20 (2H, m), 4.42–4.51 (1H, m), 5.23 (1H, t, J=9.7 Hz), 5.28–5.33 (1H, m), 5.43 (1H, t, J=9.4 Hz), 5.84 (1H, d, J=8.2 Hz), 6.74–6.78 (2H, m), 7.01–7.04 (2H, m). 13C-NMR (CDCl3) δ: 13.19 (q), 22.05 (q), 22.10 (q), 23.22 (q), 26.23 (t), 26.88 (q×3), 27.03 (q×6), 27.15 (q×3), 38.66 (s), 38.74 (s), 38.77 (s), 38.83 (s), 61.61 (t), 67.64 (d), 69.83 (d), 70.71 (d), 72.21 (d), 72.89 (d), 97.00 (d), 111.52 (s), 115.96 (d×2), 129.17 (d×2), 131.23 (s), 141.82 (s), 156.30 (s), 160.57 (s), 170.88 (s), 176.38 (s), 176.40 (s), 177.20 (s), 178.03 (s). HR-MS (ESI) m/z: 787.4373 [M+H]+ (Calcd for C42H63N2O12: 787.4376).
4-[(4-Isopropoxyphenyl)methyl]-5-methyl-3-(2,3,4,6-tetra-O-pivaloyl-β-D-glucopyranosyloxy)-1H-pyrazole (2b)A mixture of 11c (0.787 g, 1.0 mmol) and KHCO3 (0.030 g, 0.30 mmol) in MeOH (3 mL) was stirred at 50°C for 1 h; AcOH (0.018 g, 0.30 mmol) was then added at room temperature. After the reaction mixture had been concentrated under reduced pressure, the residue was purified by silica gel chromatography (eluent AcOEt–n-hexane, 1 : 2) to provide 2b (0.731 g, 98% yield). An analytical sample of 2b was obtained as a white solid by recrystallization from 2-PrOH. mp 159–163°C. [α]D20 −10.8 (c=1.0, DMSO). IR (KBr) cm−1: 3383, 2975, 2936, 2874, 1746, 1729, 1507, 1482. 1H-NMR (CDCl3) δ: 1.05 (9H, s), 1.12 (9H, s), 1.15 (9H, s), 1.18 (9H, s), 1.30 (6H, dd, J=1.2, 6.0 Hz), 2.06 (3H, s), 3.54 (2H, s), 3.84–3.88 (1H, m), 4.11–4.20 (2H, m), 4.41–4.53 (2H, m), 5.23–5.32 (2H, m), 5.38 (1H, t, J=9.2 Hz), 5.68 (2H, d, J=8.0 Hz), 6.75 (2H, d, J=8.8 Hz), 7.04 (2H, d, J=8.8 Hz), 8.82 (1H, br s). 13C-NMR (CDCl3) δ: 10.42 (q), 22.07 (q), 22.11 (q), 22.6 (t), 26.95 (q×3), 27.04 (q×3), 27.06 (q×3), 27.14 (q×3), 38.68 (s), 38.72 (s), 38.74 (s), 38.83 (s), 61.53 (t), 67.57 (d), 69.84 (d), 70.94 (d), 72.41 (d), 72.45 (d), 97.72 (d), 103.62 (s), 115.80 (d×2), 129.23 (d×2), 132.68 (s), 138.00 (s), 156.00 (s), 159.83 (s), 176.36 (s), 176.55 (s), 177.24 (s), 178.12 (s). HR-MS (ESI) m/z: 745.4262 [M+H]+ (Calcd for C40H61N2O11: 745.4270).
General Procedure for the Preparation of 4-(Substituted benzyl)-5-methyl-3-(2,3,4,6-tetra-O-pivaloyl-β-D-glucopyranosyloxy)-1H-pyrazoles (2b–d)Acetic anhydride (0.325 g, 3.18 mmol) was added dropwise to a mixture of an appropriate 1,2-dihydro-4-(substituted benzyl)-5-methyl-3H-pyrazol-3-one (3a–c) (3.0 mmol), K2CO3 (0.456 g, 3.3 mmol) in DMF (2 mL) at 40°C. The mixture was stirred for 30 min at 40°C and for 1 h at 70°C. The reaction mixture was cooled to 40°C and MeCN (25 mL), and an aqueous solution of 25% K2CO3 (3.33 g, 6.0 mmol) and 5b (2.26 g, 3.9 mmol) were added successively to the mixture. The mixture was stirred for 6 h at 60°C and cooled to room temperature. AcOEt (25 mL) and H2O (5 mL) were added to the mixture with stirring and the aqueous layer was removed. The organic layer was concentrated under reduced pressure. KHCO3 (0.090 g, 0.90 mmol) was added to a solution of the residue in MeOH (10 mL) and the mixture was stirred for 2 h at 60°C. After the addition of AcOH (0.054 g, 0.90 mmol), the reaction mixture was concentrated under reduced pressure. The residue was purified by silica gel chromatography (eluent AcOEt–n-hexane, 1 : 3) to provide 2b–d.
4-[(3-Fluoro-4-methoxyphenyl)methyl]-5-methyl-3-(2,3,4,6-tetra-O-pivaloyl-β-D-glucopyranosyloxy)-1H-pyrazole (2c)An analytical sample of 2c was obtained as a white solid by recrystallization from 2-PrOH–n-hexane. mp 189–192°C. [α]D20 −11.1 (c=1.0, DMSO). IR (KBr) cm−1: 3373, 2970, 2909, 2874, 1747, 1729, 1603, 1516, 1482, 1396, 1365. 1H-NMR (CDCl3) δ: 1.05 (9H, s), 1.12 (9H, s), 1.15 (9H, s), 1.18 (9H, s), 2.07 (3H, s), 3.54 (2H, s), 3.84 (3H, s), 3.84–3.89 (1H, m), 4.11–4.21 (2H, m), 5.23–5.31 (2H, m), 5.39 (1H, t, J=9.5 Hz), 5.69 (1H, d, J=8.0 Hz), 6.81–6.89 (3H, m), 8.93 (1H, br s). 13C-NMR (CDCl3) δ: 10.36 (q), 26.49 (t), 26.92 (q×3), 27.03 (q×6), 27.14 (q×3), 38.68 (s), 38.72 (s), 38.74 (s), 38.83 (s), 56.32 (q), 61.50 (t), 67.56 (d), 70.93 (d), 72.35 (d), 72.48 (d), 97.76 (d), 102.97 (s), 113.36 (d, JCF=2.2 Hz), 115.88 (d, JCF=18.3 Hz), 123.76 (d, JCF=2.9 Hz), 133.89 (d, JCF=5.9 Hz), 138.04 (s), 145.65 (d, JCF=10.3 Hz), 152.23 (d, JCF=244.9 Hz), 159.75 (s), 176.37 (s), 176.59 (s), 177.25 (s), 178.11 (s). HR-MS (ESI) m/z: 735.3844 [M+H]+ (Calcd for C38H56FN2O11: 735.3863).
4-[(2-Fluoro-4-methoxyphenyl)methyl]-5-methyl-3-(2,3,4,6-tetra-O-pivaloyl-β-D-glucopyranosyloxy)-1H-pyrazole (2d)An analytical sample of 2d was obtained as a white solid by recrystallization from 2-PrOH–n-hexane. mp 167–169°C. [α]D20 −7.1 (c=1.0, DMSO). IR (KBr) cm−1: 3381, 2976, 2934, 2910, 1747, 1729, 1626, 1587, 1506, 1482, 1443, 1431, 1394, 1364. 1H-NMR (CDCl3) δ: 1.05 (9H, s), 1.13 (9H, s), 1.15 (9H, s), 1.18 (9H, s), 2.10 (3H, s), 3.54 (2H, dd, J=15.6, 20.7 Hz), 3.75 (3H, s), 3.84–3.88 (1H, m), 4.12 (1H, dd, J=4.5, 12.3 Hz), 4.17 (1H, dd, J=1.8, 12.3 Hz), 5.23–5.32 (2H, m), 5.38 (1H, t, J=9.5 Hz), 5.68 (1H, d, J=8.0 Hz), 6.52–6.59 (2H, m), 7.07 (1H, t, J=8.7 Hz), 8.93 (1H, br s). 13C-NMR (CDCl3) δ: 10.14 (q), 19.70 (d, JCF=3.3 Hz), 26.94 (q×3), 27.04 (q×6), 27.16 (q×3), 38.69 (s), 38.73 (s), 38.75 (s), 38.84 (s), 55.47 (q), 61.52 (t), 67.60 (d), 70.98 (d), 72.43 (d), 72.47 (d), 97.72 (d), 101.28 (d, JCF=25.9 Hz), 102.34 (s), 109.65 (d, JCF=2.8 Hz), 119.27 (d, JCF=16.2 Hz), 131.17 (d, JCF=6.5 Hz), 138.08 (s), 159.00 (s), 159.09 (s), 160.98 (d, JCF=244.3 Hz), 176.37 (s), 176.58 (s), 177.25 (s), 178.10 (s). HR-MS (ESI) m/z: 735.3856 [M+H]+ (Calcd for C38H56FN2O11: 735.3863).
4-[(4-Isopropoxyphenyl)methyl]-1-isopropyl-5-methyl-3-(2,3,4,6-tetra-O-pivaloyl-β-D-glucopyranosyloxy)-1H-pyrazole (15)A solution of 2b (50 g, 0.0671 mol) and 2-iodopropane (45.7 g, 0.269 mol) in DMI (105 g) was added dropwise to a suspension of NaH (8.05 g, 0.201 mol, 60% oil dispersion) in DMI (150 g) while maintaining the internal temperature between 10 and 20°C. The reaction mixture was stirred for 0.5–1 h at this temperature. The reaction mixture was added dropwise to a mixture of H2O (200 g), glacial acetic acid (8.05 g, 0.134 mol) and toluene (200 g) at 0–30°C, and the layers were separated. The organic layer was washed twice with 1% brine (300 g) and concentrated under reduced pressure. The residue was dissolved in 2-PrOH (250 g) and the solution was concentrated under reduced pressure. The residue was dissolved in 2-PrOH and adjusted to a final weight of 250 g. The 2-propanol solution was stirred at 20°C for 2 h and the resulting slurry was stirred at −8~−2°C for an additional 2 h. The slurry was filtered and the wet cake washed with 2-PrOH (50 g), which was cooled to 0°C. The precipitate was dried in vacuo at 60°C to give 45.4 g (86% yield) of 15 as a white solid. mp 137–138°C. [α]D20 −2.2 (c=1.0, DMSO). IR (KBr) cm−1: 2979, 2940, 1747, 1508, 1483, 1398, 1384, 1370. 1H-NMR (CDCl3) δ: 1.04 (9H, s), 1.13 (9H, s), 1.15 (9H, s), 1.19 (9H, s), 1.29 (6H, d, J=6.3 Hz), 1.33 (6H, dd, J=6.8, 9.3 Hz), 2.04 (3H, s), 3.52 (2H, dd, J=15.8, 19.6 Hz), 3.81–3.85 (1H, m), 4.10–4.19 (2H, m), 4.18–4.28 (1H, m), 4.41–4.51 (1H, m), 5.21–5.30 (2H, m), 5.41 (1H, t, J=9.6 Hz), 5.74 (1H, d, J=8.3 Hz), 6.74 (2H, d, J=8.7 Hz), 7.04 (2H, d, J=8.7 Hz). 13C-NMR (CDCl3) δ: 9.67 (q), 22.10 (q), 22.12 (q), 22.23 (q), 22.24 (q), 26.90 (t), 26.94 (q×3), 27.05 (q×3), 27.09 (q×3), 27.16 (q×3), 38.68 (s), 38.72 (s), 38.74 (s), 38.84 (s), 49.16 (d), 61.72 (t), 67.72 (d), 69.81 (d), 71.13 (d), 72.49(d), 72.51 (d), 98.02 (d), 102.89 (s), 115.71 (d×2), 129.18 (d×2), 133.43 (s), 135.72 (s), 155.88 (s), 157.69 (s), 176.38 (s), 176.58 (s), 177.27 (s), 178.18 (s). HR-MS (ESI) m/z: 787.4740 [M+H]+ (Calcd for C43H67N2O11: 787.4739).
3-(β-D-Glucopyranosyloxy)-4-[(4-isopropoxyphenyl)methyl]-1-isopropyl-5-methyl-1H-pyrazole (1a)A methanolic solution of 28% MeONa (1.93 g, 10 mmol) was added to a suspension of 15 (7.87 g, 10 mmol) in MeOH (75 mL) at room temperature. The mixture was then heated to 55°C and stirred for 3 h at this temperature. After cooling to 40°C, acetic acid (0.601 g, 10 mmol) was added dropwise to the reaction mixture. The reaction mixture was concentrated under reduced pressure to evaporate the methyl pivalate contained in the mixture. The residue was purified by silica gel chromatography (eluent dichloromethane–MeOH, 10 : 1) to provide 1a (4.45 g, 99% yield) as a pale yellowish oil. [α]D20 −8.1 (c=1.0, DMSO). IR (KBr) cm−1: 3407, 2975, 2931, 1506, 1466, 1384. 1H-NMR (CD3OD) δ: 1.26 (6H, d, J=6.0 Hz), 1.36 (6H, dd, J=3.8, 6.8 Hz), 2.09 (3H, s), 3.21–3.26 (1H, m), 3.33–3.43 (3H, m), 3.62–3.72 (3H, m), 3.77 (1H, dd, J=2.5, 12.1 Hz), 4.36–4.46 (1H, m), 4.46–4.55 (1H, m), 5.00–5.05 (1H, m), 6.76 (2H, d, J=8.7 Hz), 7.07 (2H, d, J=8.7 Hz). 13C-NMR (CD3OD) δ: 8.93 (q), 21.61 (q×2), 21.62 (q), 21.65 (q), 26.79 (t), 49.77 (d), 61.85 (t), 70.25 (d), 70.48 (d), 74.32 (d), 77.24 (d), 77.49 (d), 102.41 (d), 104.50 (s), 116.18 (d×2), 129.39 (d×2), 134.00 (s), 137.53 (s), 156.55 (s), 159.47 (s). HR-MS (ESI) m/z: 451.2444 [M+H]+ (Calcd for C23H35N2O7: 451.2439).
5-Methyl-4-[4-(1-methylethoxy)benzyl]-1-(1-methylethyl)-1H-pyrazol-3-yl-6-O-(ethoxycarbonyl)-β-D-glucopyranoside Ethanolate (16)A solution of ethyl chloroformate (522 mg, 4.81 mmol) in MeCN (1 mL) was added dropwise to a mixture of 1a (1.89 g, 4.19 mmol), 2,6-lutidine (672 mg, 6.28 mmol) and pyridine (13 mg, 0.17 mmol) in MeCN (10 mL) while maintaining the temperature between −3 and 3°C. The reaction mixture was stirred at 0°C for 2 h. After addition of glacial acetic acid (113 mg, 1.88 mmol), the reaction mixture was allowed to warm to room temperature. The reaction mixture was diluted with MTBE (10 mL) and 10% brine (5 mL), and then the layers were separated. The organic layer was washed twice with brine (5 mL), dried over anhydrous MgSO4 (2 g) and concentrated under reduced pressure. The residue was dissolved in EtOH (17 mL) and concentrated again under reduced pressure. EtOH was added to the residue, and the weight was adjusted to 9.3 g. To the EtOH solution, n-heptane (6 mL) was added and heated to 60°C to dissolve solids. The mixture was cooled to 45°C and stirred for 1 h at this temperature for an additional 1 h at 0–5°C. The slurry was filtered and the wet cake washed successively with a mixed solvent of EtOH (1.2 mL) and n-heptane (2.8 mL), which was cooled to 0°C, and then n-heptane (2.8 mL). The precipitate was dried in vacuo at room temperature to give 1.72 g (72% yield) of 16 as a white solid. mp 70–74°C. [α]D20 −17.7 (c=1.0, DMSO). IR (KBr) cm−1: 3353, 2980, 2926, 1753, 1731, 1508, 1477, 1467, 1449, 1386, 1371. 1H-NMR (CDCl3) δ: 1.23 (3H, t, J=7.0 Hz), 1.28 (3H, t, J=7.0 Hz), 1.30 (6H, d, J=6.0 Hz), 1.38 (6H, dd, J=2.3, 6.6 Hz), 2.06 (3H, s), 3.47–3.63 (6H, m), 3.71 (2H, q, J=7.0 Hz), 4.17 (2H, q, J=7.0 Hz), 4.24–4.31 (1H, m), 4.32–4.39 (2H, m), 4.43–4.52 (1H, m), 4.98 (1H, d, J=7.6 Hz), 6.77 (2H, d, J=8.6 Hz), 7.05 (2H, d, J=8.6 Hz). 13C-NMR (CDCl3) δ: 9.72, 14.21, 18.35, 22.09, 22.21, 22.25, 26.87, 49.44, 58.35, 64.23, 66.48, 69.49, 69.86, 73.65, 74.24, 76.44, 102.32, 104.67, 115.78, 129.10, 133.15, 136.55, 155.46, 155.96, 158.07. HR-MS (ESI) m/z: 523.2648 [M+H]+ (Calcd for C26H39N2O9: 523.2650).
5-Methyl-4-[4-(1-methylethoxy)benzyl]-1-(1-methylethyl)-1H-pyrazol-3-yl-6-O-(ethoxycarbonyl)-β-D-glucopyranoside (1b)16 (1.50 g, 2.64 mmol) was dissolved in MTBE (10 mL) at 45°C The solution was concentrated under reduced pressure to evaporate EtOH. MTBE was added to the residue, and the weight was adjusted to 9.0 g. H2O (0.015 mL) and n-heptane (3.6 g) were added to the solution at 40°C and the solution was cooled to 25°C. The solution was seeded with 1a and stirred at 25°C for 3 h. The resulting slurry was warmed to 40°C, and then a mixed solvent of MTBE (0.44 g) and n-heptane (2.4 g) was added dropwise to the slurry while maintaining the temperature between 37 and 43°C. The slurry was stirred at 40°C for 1 h and for an additional 3 h at 10°C. The slurry was filtered and the wet cake washed successively with a mixed solvent of MTBE (1.5 g) and n-heptane (1.5 g) followed by n-heptane (3.0 g). The product was dried in vacuo at room temperature to give 1.35 g (98% yield) of 1a as a white solid. mp 80–83°C. [α]D20 −19.3 (c=1.0, DMSO). IR (KBr) cm−1: 3414, 2979, 1747, 1506, 1477, 1474, 1466, 1458, 1449, 1382, 1370, 1317. 1H-NMR (CD3OD) δ: 1.23 (3H, t, J=7.2 Hz), 1.26 (6H, d, J=6.1 Hz), 1.37 (6H, dd, J=2.3, 6.7 Hz), 2.07 (3H, s), 3.34–3.42 (4H, m), 3.61–3.69 (2H, m), 4.12 (2H, q, J=7.2 Hz), 4.21 (1H, dd, J=5.4, 11.5 Hz), 4.35 (1H, dd, J=1.7, 11.6 Hz), 4.35–4.45 (1H, m), 4.45–4.54 (1H, m), 5.04–5.06 (1H, m), 6.75 (2H, d, J=8.6 Hz), 7.06 (2H, d, J=8.6 Hz). 13C-NMR (CD3OD) δ: 9.70, 14.60, 22.43, 22.49, 22.54, 27.63, 50.53, 65.07, 67.67, 71.07, 71.21, 75.02, 75.56, 77.84, 103.25, 105.62, 116.98, 130.21, 134.81, 138.21, 156.65, 157.33, 159.99. HR-MS (ESI) m/z: 523.2651 [M+H]+ (Calcd for C26H39N2O9: 523.2650).