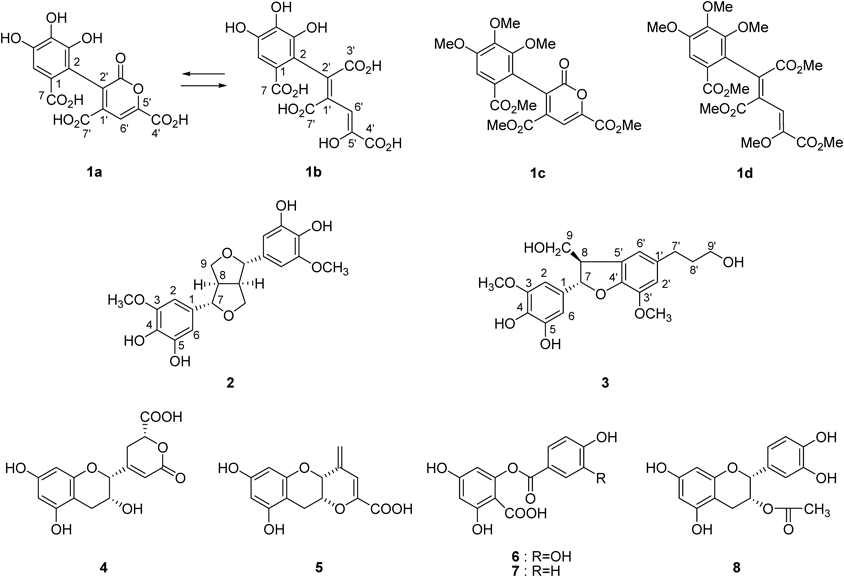
Results and Discussion
Commercial ripe tea produced in Menghai, Yunnan, China, and Japanese green tea was pulverized with water and stored at 37°C for 5 d. The aqueous acetone extract was separated by repeated column chromatography using Sephadex LH-20, Diaion HP20SS, Chromatorex ODS, and Toyopearl Butyl-650C to give three new compounds 1–3, together with 24 known compounds (Fig. 1, Supplementary Fig. S2). Based on a comparison of spectroscopic data, the known compounds were identified to be 4-[(2R,3R)-3,4-dihydro-3,5,7-trihydroxy-2H-1-benzopyran-2-yl]-3,6-dihydro-6-oxo-(2R)-2H-pyran-2-carboxylic acid (4),10) teadenol A (5),11) 2-(3,4-dihydroxybenzoyloxy)-4,6-dihydroxybenzoic acid (6),13) 2,4-dihydroxy-6-(4-hydroxybenzoyloxy)-benzoic acid (7),13) 3-O-acetyl-(−)-epicatechin (8),14) epiafzelechin (9),15) cis-epigallocatechin-3-O-p-coumarate (10),16) trans-epigallocatechin-3-O-p-coumarate (11),16) procyanidin B4 (12),17) epiafzelechin-(4β→8)-epicatechin-3-O-gallate (13),18) epiafzelechin-(4β→8)-epicatechin (14),19) procyanidin B-5 (15),20) epiafzelechin-(4β→6)-epicatechin-3-O-gallate (16),18) luteolin (17),21) tricetin (18),22) apigenin 6-C-β-glucopyranoside (19),23) kaempferol (20),24) quercetin (21),24) myricetin (22),25) dihydromyricetin (23),26) (7R*,8S*)-4,7,9,9′-tetrahydroxy-3,3′-dimethoxy-8-O-4′-neolignan (24),27) 4-hydroxybenzoic acid (25), 4-hydroxybenzoic acid methyl ester (26), and p-coumaric acid (27). In this study, four major tea catechins were not purified. Among the isolated compounds, 4 and 5 have been isolated from commercial fermented tea,10,11) while metabolites 6 and 7 are known to be microbial degradation products of quercetin and kaempferol, respectively.28) In addition, compound 8, which has only been reported as a synthetic compound,14) was isolated from tea products for the first time.
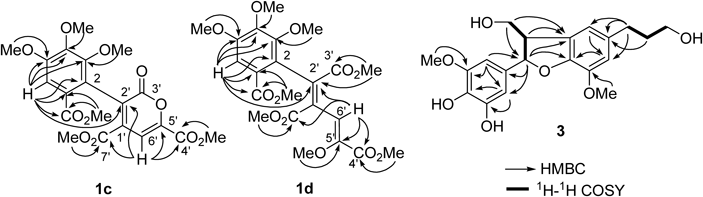
Compound 1 is a yellow pigment and was isolated as a dark brown microcrystalline powder, mp 224–226°C. The UV spectrum showed broad absorptions at 400 nm. In addition to the results of elemental analysis, the [M]− peak at m/z 352 in FAB-MS (negative mode) and the [M]+ peak at m/z 352 in electrospray ionization-time of flight (ESI-TOF)-MS suggested the molecular formula, C14H8O11. The 1H-NMR spectrum (Table 1) showed only 4 singlet signals attributable to sp2 protons, and the 13C-NMR spectrum showed complex signals arising from major (1a) and minor (1b) components. The signals of the major component 1a suggested presence of a gallic acid moiety (δC 121.4, 116.4, 144.6, 139.0, 146.4, 111.3, 169.7), three conjugated carboxyl groups (δC 163.1, 162.1, 166.0), and two different double bonds (δC 139.6, 134.7, 148.1, 111.1) including a methine (δC 111.1) and an oxygenated (δC 148.1) carbon. The signals of the minor component were also assignable to related carbons, suggesting that 1 exists as an equilibrium mixture. Methylation of 1 with diazomethane yielded an inseparable mixture of derivatives 1c and d. ESI-TOF-MS showed two [M+Na]+ peaks at m/z 459 and 505, indicating that the major derivative 1c is hexamethylate and the minor derivative 1d is octamethylate. In the heteronuclear multiple bond connectivity (HMBC) spectrum, correlations of four methoxy protons of 1c [δH 3.70 (3-OMe), 3.91 (4-OMe), 3.93 (5-OMe), 3.75 (7-OMe)] and an aromatic singlet [δH 7.42 (H-6)] confirmed the presence of the gallic acid moiety (Fig. 2). An olefinic singlet at δH 7.53 (H-6′) showed HMBC correlations with two carboxyl carbons at δC 163.8 (C-7′) and 159.7 (C-4′), and with two olefinic carbons at δC 133.3 (C-2′) and 146.6 (C-5′). The carbon signal at δC 133.3 (C-2′) also showed a 4J correlation with the H-6 of the gallic acid unit. The carboxy carbon signal at δC 160.4 (C-3′) did not show HMBC correlation, and it was deduced that this carboxy carbon forms a lactone ring with the oxygen atom at C-5′. From these spectroscopic observations, structure 1c was deduced to be as shown in Fig. 1. The HMBC correlations of the other methylate 1d showed the presence of three methoxycarbonyl groups in addition to that of the gallic acid unit. Furthermore, an enol methyl ether structure was confirmed by correlations of the methyl group at δH 3.62 (5′-OMe) and an olefinic proton at δH 7.90 (H-6′) to C-5′ (δC 148.0) (Fig. 2). Based on these spectroscopic and chemical results, the structure of yellow pigment 1 was determined as shown in Fig. 1 and named theagalloflavic acid.
Table 1.
1H- (500 MHz) and
13C- (125 MHz) NMR Date for
1a–
d| Position | 1aa) | 1ba) | 1cb) | 1db) |
|---|
| δH | δC | δH | δC | δH | δC | δH | δC |
|---|
| 1 | — | 121.4 | — | 121.2 | — | 124.0 | — | 125.7 |
| 2 | — | 116.4 | — | 116.8 | — | 122.5 | — | 124.5 |
| 3 | — | 144.6 | — | 144.6 | — | 150.7 | — | 151.9 |
| 4 | — | 139.0 | — | 139.0 | — | 145.6 | — | 145.0 |
| 5 | — | 146.4 | — | 146.3 | — | 153.5 | — | 153.1 |
| 6 | 7.21 (1H, s) | 111.3 | 7.21 (1H, s) | 111.5 | 7.42 (1H, s) | 109.5 | 7.24 (1H, s) | 108.6 |
| 7 | — | 169.7 | — | 169.7 | — | 165.8 | — | 166.3 |
| 1′ | — | 139.6 | — | 139.6 | — | 137.6 | — | 137.4 |
| 2′ | — | 134.7 | — | 134.5 | — | 133.3 | — | 131.2 |
| 3′ | — | 163.1 | — | 163.1 | — | 160.4 | — | 165.9 |
| 4′ | — | 162.1 | — | 162.1 | — | 159.7 | — | 163.8 |
| 5′ | — | 148.1 | — | 148.1 | — | 146.6 | — | 148.0 |
| 6′ | 7.44 (1H, s) | 111.1 | 7.46 (1H, s) | 111.1 | 7.53 (1H, s) | 109.9 | 7.90 (1H, s) | 117.5 |
| 7′ | — | 166.0 | — | 166.0 | — | 163.8 | — | 167.6 |
| 3-OMe | | | | | 3.70 (3H, s) | 61.1 | 3.82 (3H, s) | 60.8 |
| 4-OMe | | | | | 3.91 (3H, s) | 60.8 | 3.89 (3H, s) | 60.8 |
| 5-OMe | | | | | 3.93 (3H, s) | 56.1 | 3.90 (3H, s) | 56.0 |
| 7-OMe | | | | | 3.75 (3H, s) | 52.3 | 3.79 (3H, s) | 52.2 |
| 3′-OMe | | | | | | | 3.72 (3H, s) | 52.3 |
| 4′-OMe | | | | | 3.95 (3H, s) | 53.1 | 3.83 (3H, s) | 52.4 |
| 5′-OMe | | | | | | | 3.62 (3H, s) | 60.1 |
| 7′-OMe | | | | | 3.66 (3H, s) | 52.8 | 3.40 (3H, s) | 51.7 |
a) Measured in CD3OD. b) Measured in CDCl3.
The molecular formula of 2, C20H22O8, was confirmed by high resolution (HR)-ESI-MS, which showed the [M+Na]+ peak at m/z 413.1196 (Calcd for C20H22O8Na: 413.1212). The 1H-NMR spectrum showed signals of phenolic methoxy methyl (δH 3.83), aliphatic methine (δH 3.09, H-8, H-8′), oxygenated methylene (δH 3.82, 4.22, H-9, H-9′), and benzylic methine (δH 4.63, H-7, H-7′), which were closely related to that of pinoresinol with a symmetric structure.29) An important difference was observed in the aromatic region: two meta-coupled aromatic doublets (H-2, H-2′) indicated the presence of a 3,4-dihydroxy-5-methoxy aromatic group. This was confirmed by comparison of the chemical shifts of 1H and 13C aromatic signals with those of (+)-5′-hydroxypinoresinol.29) The coupling constant of the benzylic methine proton (4.2 Hz) was similar to that of pinoresinol (4.4 Hz), and the 13C-NMR chemical shift of C-7 and C-7′ (δC 88.3) indicated that the aromatic groups were in equatorial positions.30) Thus, 2 was determined to be 5,5′-dihydroxypinoresinol. The positive optical rotation ([α]D+12.6) suggested that the absolute configuration was the same as that of (+)-pinoresinol ([α]D+34) and (+)-5′-hydroxypinoresinol ([α]D+40).29)
The 1H- and 13C-NMR data of compound 3 were similar to those of dihydrodehydrodiconiferyl alcohol,31) and HR-ESI-MS analysis gave C20H24O7 as the molecular formula (m/z 399.1403 [M+Na]+, Calcd for C20H24O7Na: 399.1420), suggesting that 3 is an oxygenated derivative of dihydrodehydrodiconiferyl alcohol. Instead of the trisubstituted benzene ring of dihydrodehydrodiconiferyl alcohol, the presence of a tetrasubstituted benzene ring in 3 was apparent from the meta-coupled signals at δH 6.52 and 6.49 (each doublet, J=1.6 Hz, H-2, H-6), indicating that oxygenation occurred on the trisubstituted benzene ring of dihydrodehydrodiconiferyl alcohol. This was further confirmed by HMBC correlations shown in Fig. 2. The relative stereochemistry of C-7 and C-8 was deduced to be a trans-configuration from the chemical shift of C-8 (δC 55.6).32) The electron-circular dichroism spectrum showed a negative Cotton effect at 280 nm (Δε−0.24), the sign of which was opposite of the Cotton effect of the related compound (7S,8R)-4,9-dihydroxy-3,3′,5-trimethoxy-4′,7-epoxy-8,5′-neolignan-9′-oic acid methyl ester [Δε+0.13 at 282 nm],32) indicating 7R,8S configuration of 3. Thus, compound 3 was characterized as (7R,8S)-dihydro-3′-hydroxy-8-hydroxymethyl-7-(4,5-dihydroxy-3-methoxyphenyl)-1′-benzofuranpropanol.
In this study, we reproduced the initial stage of polyphenol degradation during microbial fermentation of green tea by mixing green tea with commercial Chinese ripe-tea retaining microorganisms.33,34) Compounds 2 and 3 were apparently produced by simple oxygenation of pinoresinol and dihydrodehydrodiconiferyl alcohol, respectively, during microbial fermentation. This conclusion was supported by a preceding study that reported the isolation of 5′-hydroxypinoresinol as a metabolite of pinoresinol after treatment with Aspergillus sp.22) In contrast, 1 is considered to be an oxidative metabolite of hexahydroxydiphenoyl group, which are typical acyl group of ellagitannin and also a precursor of ellagic acid (Chart 1). Tea leaves contains 1-O-galloyl-4,5-(S)-hexahydroxydiphenoyl-β-D-glucose,17) and 1 may be generated by oxidative cleavage of the aromatic ring of the hexahydroxydiphenoyl esters followed by hydrolysis. It is known that hydrolysis of the hexahydroxydiphenoyl esters affords ellagic acid; however, ellagic acid was barely detected in the extract. Therefore, 1 was probably derived from hexahydroxydiphenoyl esters directly. It may be also possible that 1 was produced by oxidative dimerization of galloyl esters, because the concentration of hexahydroxydiphenoyl esters in tea leaves is much lower than catechin galloyl esters. The mechanisms of oxidative cleavage of the aromatic ring and conjugated double bond are probably related to the formation of 4–7, and bacterial catechol dioxygenases are responsible to the oxidation.35) Five commercial ripe-tea including that used in this experiment were analyzed by HPLC, however, presence of 1 could not be confirmed. Probably, this pigment is produced only in the early stage of microbial fermentation and decomposed in the continuation of fermentation. The degradation of the polyphenols may be related to production of theabrownin,33,34) uncharacterized major polyphenols of ripe-tea detected as a broad hump on HPLC baseline. Further study on the microbial degradation of tea polyphenols is now in progress.
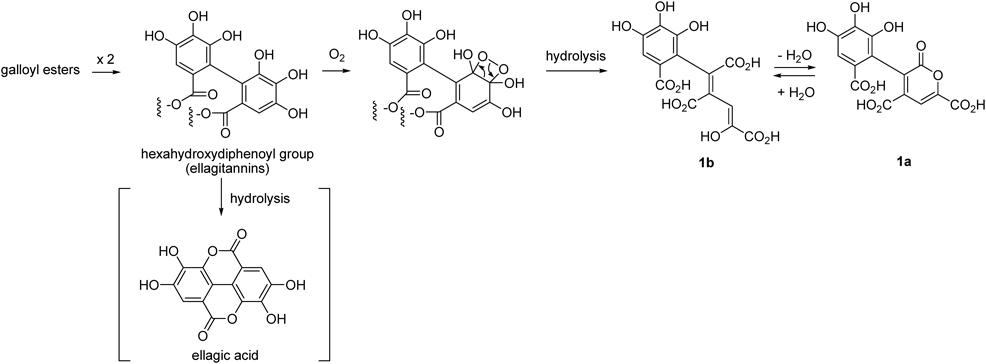
Chart 1. Possible Production Mechanism of 1
Experimental
GeneralOptical rotations were measured on a JASCO P-1020 digital polarimeter (Jasco, Tokyo, Japan). IR spectra were measured on a JASCO FT/IR 410 spectrophotometer. UV spectra were obtained on a JASCO V-560 UV/Vis spectrophotometer. The electron capture detector (ECD) spectra were measured with a JASCO J-725N spectrophotometer. 1H-NMR, 13C-NMR, 1H–1H correlation spectroscopy (COSY), heteronuclear single quantum coherence (HSQC), and HMBC spectra were measured on a Varian UNITY plus 500 (500 MHz for 1H- and 125 MHz for 13C-) NMR spectrometer (Varian, Palo Alto, CA, U.S.A.), and a JEOL JNM-AL400 (400 MHz for 1H- and 100 MHz for 13C-) NMR spectrometer (JEOL Ltd., Tokyo, Japan). Coupling constants (J) are expressed in Hertz and chemical shifts (δ) are reported in ppm with the solvent signal used as a standard (acetone-d6: δH 2.04, δC 29.8). FAB-MS data were recorded on a JMS700N spectrometer (JEOL Ltd.) using m-nitrobenzyl alcohol or glycerol as the matrix. ESI-TOF-MS were obtained on a JMS-T100TD spectrometer (JEOL Ltd.). Column chromatography was performed using Diaion HP20SS (Mitsubishi Chemical Co., Tokyo, Japan), Sephadex LH-20 (25–100 µm, GE Healthcare UK Ltd., Little Chalfont, U.K.), Chromatorex ODS (Fuji Silysia Chemical Ltd., Kasugai, Japan), and Toyopearl Butyl-650C (Tosoh Bioscience, Tokyo, Japan). Thin-layer chromatography (TLC) was performed on 0.2-mm-thick precoated Kieselgel 60 F254 plates (Merck) using toluene–ethyl formate–formic acid (1 : 7 : 1, v/v). Spots were detected by UV illumination and sprayed with 2% methanolic FeCl3. Analytical reverse-phase HPLC was performed on a Cosmosil 5C18-AR II column (Nacalai Tesque Inc.; 4.6 mm i.d.×250 mm) using an elution gradient of 4–30% (39 min) and 30–75% (15 min) CH3CN in 50 mM H3PO4 (flow rate 0.8 mL/min; detection using a JASCO photodiode array detector MD-910).
MaterialsGreen tea was produced at the Higashisonogi Tea Research Station, Nagasaki Agricultural and Forestry Technical Development Center, Nagasaki, Japan. Chinese ripe pu-erh was obtained in Meng Hai, Yunnan, China, in 2006.
Mixed Fermentation and SeparationGreen tea (200 g) was mechanically mixed with Chinese ripe pu-erh (100 g) and water (200 mL) in a Waring blender. The mixture was stored at 37°C for 5 d in an incubator with occasional mixing and spraying with water to maintain humidity. The fermented tea was extracted with 60% acetone (3 L×5). The extract was concentrated and the resulting aqueous solution (0.8 L) was partitioned with EtOAc (1.0 L×2, neutral EtOAc fraction, 23.6 g). The aqueous layer was acidified by addition of trifluoroacetic acid (pH 3) and further partitioned with EtOAc (1.0 L×2, acidic EtOAc fraction, 9.6 g). The aqueous layer was concentrated to remove EtOAc and a precipitate mainly composed of polymeric polyphenols (6.3 g) was collected by filtration. The filtrate was applied to a Diaion HP20SS column (5 cm i.d.×20 cm) with water containing increasing amounts of MeOH (10% stepwise, each 200 mL) to give five fractions: Fr. 1 (55.1 g), Fr. 2 (6.96 g, major constituent was epigallocatechin as indicated by HPLC), Fr. 3 (3.93 g, epigallocatechin, epicatechin, and procyanidin B-4), Fr. 4 (8.60 g, caffeine), and Fr. 5 (1.26 g, caffeine and tea saponins). An aqueous solution of part (44 g) of Fr. 1 was acidified with trifluoroacetic acid to pH 2 and subjected to Sephadex LH-20 column chromatography (7×20 cm) with water to give 1 (0.52 g). An acidic EtOAc fraction was separated by Diaion HP20SS column chromatography (4 cm i.d.×24 cm) with water containing increasing amounts of MeOH (10% stepwise, each 200 mL) to give eight fractions. HPLC analysis characterized the main fractions as: Fr. aA2, 0.56 g, major compound was gallic acid; Fr. aA3, 2.96 g, gallic acid, gallocatechin, and epigallocatechin; Fr. aA4, 0.87 g; Fr. aA5, 2.31 g, caffeine, epicatechin, and epigallocatechin-3-O-gallate; Fr. aA6, 1.26 g; Fr. aA7, 0.36g, caffeine and epicatechin-3-O-gallate. Fraction aA4 was loaded onto a Sephadex LH-20 column (3×27 cm) and eluted with 30–100% MeOH (10% stepwise) to give theobromine (29 mg), 4 (45 mg), 12 (90 mg), and a mixture of catechin, epicatechin, and epigallocatechin (405 mg). Fraction aA6 was treated similarly on a Sephadex LH-20 column (3×27 cm) with 60–100% MeOH (10% stepwise) to give 5 (76 mg) and 6 (51 mg). The neutral EtOAc fraction (23.0 g) was fractionated by Diaion HP20SS column chromatography (5×25 cm, 0–100% MeOH, 10% stepwise, then acetone) into seven fractions: Frs. nA1–7 (0.64, 4.13, 2.22, 1.29, 1.77, 1.03, 1.52 g, respectively). Among the fractions, Frs. nA1–4 mainly contained caffeine, gallic acid, gallocatechin, epicatechin, epigallocatechin, epigallocatechin-3-O-gallate, and epicatechin-3-O-gallate. Fraction nA5 (1.72 g) was separated by a combination of column chromatography using Sephadex LH-20, Chromatorex ODS, Toyopearl Butyl-650C, and Diaion HP20SS to give 25 (12.6 mg), 5 (9.7 mg), 9 (9.6 mg), 14 (7.3 mg), 13 (10 mg), 15 (6 mg), and 23 (15.3 mg). Fraction nA6 (1.03 g) was separated by Sephadex LH-20 column chromatography (4×25 cm, 50–100% MeOH, 10% stepwise, each 300 mL) to yield 5 (60 mg), 6 (27 mg), and 8 (19 mg). Column chromatography of Fr. nA7 (1.52 g) on Sephadex LH-20 (3×29 cm, 50–100% MeOH, 10% stepwise, each 200 mL) gave eight subfractions. Fraction nA7-2 (75 mg) was separated by Chromatorex ODS (2×20 cm, 40–100% MeOH, 10% stepwise, each 100 mL) and Diaion HP20SS (2×20 cm, 40–100% MeOH, 10% stepwise, each 80 mL) to give 24 (5 mg). Repeated chromatography of Fr. nA7-3 (79 mg) on Chromatorex ODS and Diaion HP20SS yielded 2 (5.6 mg), 3 (8.0 mg), 25 (8.0 mg), 26 (7.0 mg), and 19 (7.0 mg). Fraction nA7-4 (134 mg) was separated by Chromatorex ODS (2×20 cm, 0–100% MeOH, 10% stepwise, each 100 mL) to give 27 (50 mg), while separation of Fr. nA 7-5 with Chromatorex ODS and Diaion HP-20SS gave 6 (32 mg) and 7 (21 mg). Similar chromatographic separation afforded 17 (9.0 mg), 18 (11 mg), 20 (3.0 mg), and 10 (10 mg) from Fr. nA7-6 (56 mg); 20 (12 mg), 21 (27 mg), 22 (24 mg), and 11 (20 mg) from Fr. nA7-7 (115 mg); and 16 (4.0 mg) from Fr. nA7-8 (26 mg).
Theagalloflavic Acid (1)Dark brown crystalline powder; mp 224–226°C; UV (MeOH) λmax nm (log ε): 400 (3.70), 286 (3.64), 254 (3.68), 222 (3.97). IR νmax cm−1: 3379, 1732, 1652, 1614. 1H- and 13C-NMR (CD3OD) see Table 1. ESI-TOF-MS m/z: 352 [M]+ (1a), FAB-MS (negative mode) m/z: 352 [M]− (1a). Anal. Calcd for C14H8O111.3H2O: C, 44.77%; H, 2.84%. Found: C, 44.69%; H, 2.56%; HR-FAB-MS m/z: 351.0007 [M−H]− (Calcd for C14H7O11: 350.9988).
Methylation of 1A MeOH solution (2 mL) of 1 (60 mg) was treated with ethereal diazomethane and purified by silica gel column chromatography with hexane–acetone to yield a mixture of 1c and d (29 mg). Methyl derivatives 1c and d: yellow solid. UV λmax (MeOH) nm (log ε): 304 (3.93), 258 (4.00), 215 (4.51). IR cm−1: 3002, 2953, 1732, 1592. 1H- and 13C-NMR (CDCl3) see Table 1. FAB-MS m/z: 459 [M+Na]+ (1c), 505 [M+Na]+ (1d); HR-ESI-TOF-MS m/z: 505.13242 [M+Na]+ (Calcd for C22H26O12Na: 505.13219).
Analysis of Commercial Ripe-TeaFive commerical ripe-tea products (produced in Anhua, Hunan, China; Wuzhou, Guangxi, China; Menghai, Yunnan, China; Ya’an, Sichuan, China; Kunming, Yunnan, China), green tea (Nagasaki, Japan), and black tea (Sri Lanka) were extracted with hot water (0.5 g in 5 mL, 80°C, 10 min). After filtration with membrane filter (0.45 µm), the filtrate was analyzed by reverse-phase HPLC using a Cosmosil 5C18-PAQ column (Nacalai Tesque Inc.; 4.6 mm i.d.×250 mm) with an elution gradient of 4–30% (39 min) and 30–75% (15 min) CH3CN in 0.1% trifluoroacetic acid (flow rate 0.8 mL/min; detection using a JASCO photodiode array detector MD-910). Compound 1 showed a peak at 6.41 min (Supplementary Fig. S3).
(+)-5,5′-Dihydroxypinoresinol (2)A white amorphous powder, ESI-MS (negative) m/z: 389 [M−H]−. ESI-MS (positive) m/z: 413 [M+Na]+. HR-ESI-TOF-MS (positive) m/z: 413.1196 [M+Na]+ (Calcd for C20H22O8Na: 413.1212). IR cm−1: 3220, 1677, 1619. UV λmax (MeOH) nm (log ε): 208 (4.90). 1H-NMR (CD3OD, 400 MHz) δ: 6.50 (2H, d, J=1.8 Hz, H-2, 2′), 6.48 (2H, d, J=1.8 Hz, H-6, 6′), 4.63 (2H, d, J=4.2 Hz, H-7, 7′), 4.22 (2H, dd, J=6.8, 9.0 Hz, H-9, 9′), 3.83 (6H, s, 3-OMe, 3′-OMe), 3.82 (2H, m, H-9, 9′), 3.09 (2H, m, H-8, 8′). 13C-NMR (CD3OD, 100 MHz) δ: 149.8 (C-3, 3′), 146.6 (C-5, 5′), 134.8 (C-1, 1′), 133.2 (C-4, 4′), 107.8 (C-6, 6′), 102.6 (C-2, 2′), 88.3 (C-7, 7′), 72.7 (C-9, 9′), 56.6 (C-OMe), 55.4 (C-8, 8′). [α]20D+12.6 (c=0.1, MeOH).
(7R,8S)-Dihydro-3′-hydroxy-8-hydroxymethyl-7-(4,5-dihydroxy-3-methoxyphenyl)-1′-benzofuranpropanol (3)A white amorphous powder. ESI-MS (negative) m/z: 357 [M−H2O−H]−, 375 [M−H]–. ESI-MS (positive) m/z: 399 [M+Na]+. HR-ESI-MS (positive) m/z: 399.1403 [M+Na]+ (Calcd for C20H24O7Na: 399.1420). IR cm−1: 3368, 1608, 1515. UV λmax (MeOH) nm (log ε): 254 (2.34), 208 (2.73). 1H-NMR (CD3OD, 500 MHz) δ: 6.71 (2H, br s, H-2′, 6′), 6.52 (1H, d, J=1.6 Hz, H-2), 6.49 (1H, d, J=1.6 Hz, H-6), 5.43 (1H, d, J=6.1 Hz, H-7), 3.84 (3H, s, 3′-OMe), 3.82 (1H, m, H-9), 3.80 (3H, s, 3-OMe), 3.74 (1H, m, H-9), 3.56 (2H, t, J=6.6 Hz, H-9′), 3.43 (1H, q, J=6.1 Hz, H-8), 2.61 (2H, t, J=7.6 Hz, H-7′), 1.80 (2H, m, H-8′). 13C-NMR (CD3OD, 125 MHz) δ: 149.7 (C-3), 147.5 (C-4′), 146.1 (C-5), 145.2 (C-3′), 136.9 (C-1′), 134.9 (C-4), 134.2 (C-1), 129.8 (C-5′), 118.0 (C-6′), 114.1 (C-2′), 107.3 (C-6), 104.1 (C-2), 89.0 (C-7), 65.1 (C-9), 62.2 (C-9′), 56.8 (3′-OMe), 56.6 (3-OMe), 55.6 (C-8), 35.8 (C-8′), 32.9 (C-7′). [α]D20−0.81 (c=0.1, MeOH). CD (MeOH) Δε (nm): −0.33 (260), +0.19 (273), −0.24 (280).