Results and Discussion
Lycopodium carinatum purchased in Bangkok, Thailand, was dried, powdered, and extracted with MeOH. The crude alkaloidal fraction obtained by a conventional procedure from the MeOH extract was successively purified by SiO2 and amino-silica gel column chromatography to give 26 alkaloids, six of which were new alkaloids (1, 2, 5, 7, 9, 10).
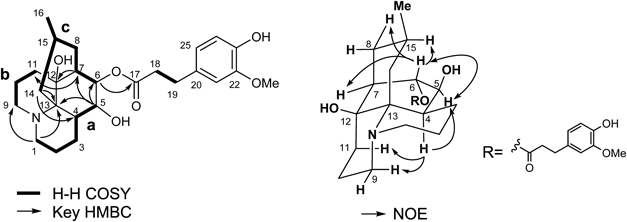
The molecular formula of new alkaloid 1, named lycopocarinamine A, was established as C26H37NO6 from high resolution-electrospray ionization (HR-ESI)-MS [m/z 460.26722 (MH+)], which possessed ten carbons more than common Lycopodium alkaloids (Fig. 1). 1H- and 13C-NMR data (Table 1) showed signals characteristic of common Lycopodium alkaloids, namely, a secondary methyl group [δH 0.88 (3H, d), δC 23.4], two sets of aminomethylenes [δH 3.24 (td, H-1), δH 3.20 (td, H-9), δH 2.47 (2H, overlapped, H-1 and 9), δC 46.6, δC 45.6], and two oxymethines [δH 4.70 (s, H-6), δH 3.47 (d, H-5), δC 82.2, δC 71.5]. Furthermore, the signals of 1,2,4-trisubstituted aromatic protons [δ 6.85 (d, J=7.7 Hz), δ 6.70 (d, J=2.1 Hz) and δ 6.69 (dd, J=7.7, 2.1 Hz)], a methoxy group [δ 3.88 (3H, s)], and two adjacent methylene protons [δ 2.90 (2H), δ 2.61 (2H)] implied the presence of a dihydroferulic acid unit.
Table 1. NMR Data for
1–
3 in CDCl
3a)| Position | Lycopocarinamine A (1) | Lycopocarinamine B (2) | Lycocarinatine A (3, revised) |
|---|
| 1H | 13C | 1H | 13C | 1H | 13C |
|---|
| 1 | 3.24 (td, 13.7, 3.4) | 46.6 | 3.25 (td, 13.7, 4.1) | 46.6 | 3.55 (td, 13.5, 3.6) | 63.3 |
| 2.47 (overlapped) | | 2.57–2.44 (overlapped) | | 2.84 (m) | |
| 2 | 1.98 (overlapped) | 18.9 | 1.95 (overlapped) | 18.8 | 1.90 (m) | 22.2 |
| 1.34 (d, 8.2) | | 1.34 (overlapped) | | 1.80 (m) | |
| 3 | 1.84 (overlapped) | 22.35 | 1.85 (overlapped) | 22.4 | 1.90 (m) | 20.3 |
| 1.41 (d, 13.1) | | 1.42 (br d, 13.1) | | 1.42 (br d, 14.0) | |
| 4 | 2.33 (br s) | 31.1 | 2.38 (br t, 8.6) | 31.1 | 2.12 (m) | 37.2 |
| 5 | 3.47 (d, 6.9) | 71.5 | 3.51 (d, 6.9) | 71.6 | 3.38 (d, 6.8) | 70.8 |
| 6 | 4.70 (s) | 82.2 | 4.73 (s) | 82.2 | 4.58 (s) | 82.1 |
| 7 | 1.79 (overlapped) | 44.1 | 1.80 (overlapped) | 44.2 | 1.73 (br s) | 45.2 |
| 8 | 1.90 (overlapped) | 33.4 | 1.92 (overlapped) | 33.4 | 2.05 (td, 12.9, 5.2) | 33.1 |
| 1.36 (t, 6.2) | | 1.36 (overlapped) | | 1.24 (m) | |
| 9 | 3.20 (td, 13.1, 3.4) | 45.6 | 3.20 (td, 12.4, 3.4) | 45.7 | 4.02 (td, 12.7, 3.3) | 59.5 |
| 2.47 (overlapped) | | 2.57–2.44 (overlapped) | | 2.95 (m) | |
| 10 | 1.98 (overlapped) | 20.5 | 2.00 (overlapped) | 20.6 | 2.84 (m) | 16.4 |
| 1.50 (d, 13.7) | | 1.52 (br d, 13.7) | | 1.52 (br d, 12.4) | |
| 11 | 1.82 (overlapped) | 30.9 | 1.87 (overlapped) | 31.0 | 1.63 (td, 14.0, 4.1) | 30.3 |
| 1.18 (d, 13.7) | | 1.21 (br d, 13.7) | | 1.24 (m) | |
| 12 | | 69.7 | | 69.7 | | 71.1 |
| 13 | | 58.4 | | 58.3 | | 72.9 |
| 14 | 2.24 (dd, 13.7, 6.2) | 35.3 | 2.25 (dd, 13.1, 6.2) | 35.4 | 2.71 (t, 12.7) | 27.5 |
| 1.18 (d, 13.7) | | 1.24 (br d, 11.7) | | 1.80 (m) | |
| 15 | 2.53 (m) | 22.26 | 2.57–2.44 (overlapped) | 22.3 | 2.58 (m) | 22.6 |
| 16 | 0.88 (3H, d, 6.2) | 23.4 | 0.88 (3H, d, 6.2) | 23.4 | 0.94 (3H, d, 6.3) | 23.2 |
| 17 | | 172.5 | | 172.4 | | 173.0 |
| 18 | 2.61 (2H, m) | 36.6 | 2.63 (2H, m) | 36.4 | 2.64 (2H, m) | 36.6 |
| 19 | 2.90 (2H, m) | 31.1 | 2.91 (2H, m) | 30.8 | 2.95 (m) | 31.6 |
| | | | | 2.84 (m) | |
| 20 | | 131.9 | | 132.6 | | 131.6 |
| 21 | 6.70 (d, 2.1) | 111.0 | 6.73 (d, 1.4) | 111.6 | 6.71 (d, 1.2) | 111.3 |
| 22 | | 146.5 | | 148.9 | | 146.6 |
| 23 | | 144.1 | | 147.6 | | 144.3 |
| 24 | 6.85 (d, 7.7) | 114.3 | 6.80 (d, 7.6) | 111.1 | 6.84 (d, 7.7) | 114.3 |
| 25 | 6.69 (dd, 7.7, 2.1) | 120.9 | 6.74 (dd, 7.6, 1.4) | 120.2 | 6.69 (dd, 7.7, 1.2) | 121.2 |
| 26 | 3.88 (3H, s) | 55.9 | 3.88 (3H, s) | 55.9 | 3.89 (3H, s) | 56.0 |
| 27 | | | 3.86 (3H, s) | 55.9 | | |
a) Measured at 600 and 150 MHz, respectively.
1H–1H correlation spectroscopy (COSY) and 1H-detected heteronuclear multiple quantum coherence (HMQC) analyses (Fig. 1) indicated the presence of three carbon chains (a: –CH2CH2CH2CHCH–; b: –CH2CH2CH2–; c: –CHCHCH2CHMeCH2–), and the gross structure of 1 was established by heteronuclear multiple bond connectivity (HMBC) analyses as follows. HMBC cross peaks between H-5 (δ 3.47) and the carbons of fragment c (δ 44.1, C-7 and δ 82.2, C-6) showed that fragment c was attached to C-5 position. The cross peak between H-7 and the terminal carbon of fragment b (δ 30.9, C-11) and a quaternary carbon (δ 69.7) indicated that fragment b was attached to C-7 via the quaternary carbon (δ 69.7). HMBC correlations between H-5 and another quaternary carbon (δ 58.4), and between H-14 (δ 2.24) and C-4 and the same quaternary carbon (δ 58.4) implied that C-14 and C-4 were connected via the quaternary carbon (δ 58.4, C-13). Correlations from H-1 to C-9 and C-13 indicated that C-1, C-9, and C-13 were connected to each other via a nitrogen atom. HMBC correlation between H-6 (δ 4.70) and an ester carbonyl carbon showed that the dihydroferulic acid unit was connected to C-6 position.
The stereochemistry of 1 was elucidated as follows. Nuclear Overhauser effect (NOE) correlations of H-4/H-9 and H-4/H-11 indicated that H-4 had an axial orientation and the stereochemistry of C-12 quaternary carbon was established, as shown in Fig. 1. NOE correlations of H-4/H-5 and H-5/H-6 implied that H-4 and H-5 were in syn orientation. The coupling pattern of H-6 (s) suggested that the dihedral angles between H-5 and H-6, and between H-6 and H-7 were respectively ca. 90°, as depicted in Fig. 1. NOE correlations of H-6/H-15 demonstrated that the methyl group was in an equatorial position. From these analyses, the structure of 1 was elucidated as shown in Fig. 1. Lycopocarinamine A (1) is the second example of a lycopodine-type alkaloid having a 5,6,12-tri-oxygenated skeleton.
The molecular formula of new alkaloid 2, named lycopocarinamine B, was deduced from HR-ESI-MS experiments to be C27H39NO6 [m/z 474.28222 (MH+)], which had an extra CH2 compared to 1. The 1H-NMR data of 2 were very similar to those of 1 except for the presence of an additional methoxy signal at δ 3.86 (3H, s). Taking these facts into consideration, 2 was presumed to be an O-methyl derivative of 1. To confirm the structure inferred by spectroscopic analyses above, the chemical conversion of 1 was attempted (Chart 1). 1 was treated with freshly prepared CH2N2 solution to afford 2. 1H-NMR data of the methylated compound was completely identical with those of natural 2 and therefore, the structure of 2 was established as shown.
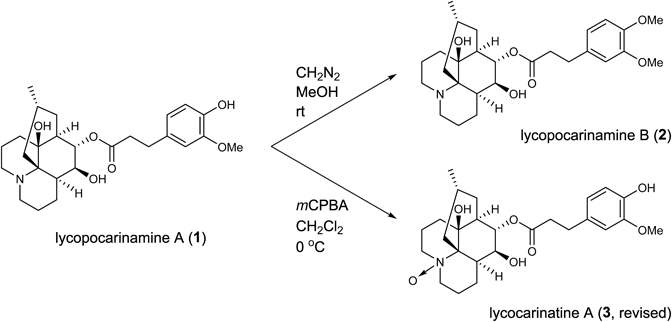
Chart 1. Chemical Conversions of Lycopocarinamine A (1)
The molecular formula of alkaloid 3 was established as C26H37NO7 from the protonated molecular ion peak [m/z 476.26125 (MH+)] in the HR-ESI-MS spectrum, which had an extra oxygen atom compared to 1. The 1H- and 13C-NMR data of 3 resembled those of 1 except for the signals around the nitrogen atom (H2-1, 9 and C-1, 9, 13). From these data, alkaloid 3 was deduced to be an N-oxide derivative of 1. To confirm the structure of 3, a chemical conversion from 1 was executed. 1 was treated with m-chloroperbenzoic acid (mCPBA) in CH2Cl2 at 0°C to give 3 in a quantitative yield. The spectral data of the oxidized compound were completely identical with those of the natural product and therefore, the structure of 3 was confirmed.
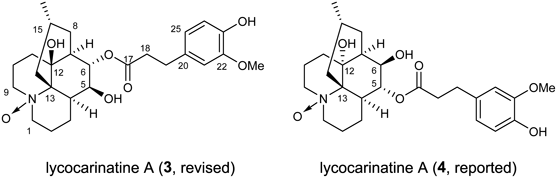
We noticed that the spectral data of 3 were in good agreement with those of recently reported lycocarinatine A (4, reported)28) (Fig. 2). In that study, the authors observed HMBC correlations between H-5 and C-12, and between H-6 and C-13. However, as those were four-bond correlations, the authors must have erred in the assignment of the signals of [H-5, C-5] for [H-6, C-6]. We hereby propose the revised structure of lycocarinatine A as shown in Fig. 2.
The HR-ESI-MS spectrum of new alkaloid 5, named lycopocarinamine C, gave a protonated molecular ion peak at m/z 458.25452 ([MH]+) that corresponded to the molecular formula C26H35NO6 (Fig. 3). 1H-NMR data (Table 2) showed signals characteristic of Lycopodium alkaloids, namely, a secondary methyl group [δ 1.00 (3H, d)], two sets of aminomethylenes [δ 2.93 (m, H-1) and δ 2.53 (m, H-1), δ 2.62 (2H, m, H2-9)], and an olefinic proton [δ 5.40 (t, H-11)]. Furthermore, the signals of 1,2,4-trisubstituted aromatic protons [δ 6.68 (dd, J=8.2, 2.1 Hz), δ 6.82 (d, J=8.2 Hz), and δ 6.68 (d, J=2.1 Hz)], a methoxy group [δ 3.88 (3H, s)], and two adjacent methylene protons [δ 2.85 (2H), δ 2.58 (2H)] indicated the presence of a dihydroferulic acid unit. The 1H- and 13C-NMR data highly resembled those of known Lycopodium alkaloid malycorin B (6).29) The only differences between 5 and 6 were the absence of an acetyl group in 5 and the chemical shift of proton at C-8 (5; δ 3.30, 6; δ 4.46). Judging from these spectroscopic data, 5 was deduced to be an 8-O-deacetyl derivative of malycorin B (6).
Table 2. NMR Data for
5 and
6 in CDCl
3a)| Position | Lycopocarinamine C (5) | Malycorin B (6) |
|---|
| 1H | 13C | 1H | 13C |
|---|
| 1 | 2.93 (m) | 48.5 | 2.94 (m) | 48.4 |
| 2.53 (m) | | 2.54 (m) | |
| 2 | 1.77 (m) | 24.9 | 1.76 (m) | 24.8 |
| 1.64 (m) | | 1.63 (br s) | |
| 3 | 1.77 (m) | 22.5 | 1.85 (br s) | 22.6 |
| 1.42 (br d, 8.9) | | 1.42 (d, 8.8) | |
| 4 | 1.95 (m) | 43.9 | 1.95 (m) | 43.7 |
| 5 | 3.60 (d, 6.2) | 71.1 | 3.62 (br s) | 71.4 |
| 6 | 5.13 (s) | 75.1 | 4.96 (m) | 75.0 |
| 7 | 2.53 (m) | 51.2 | 2.67 (m) | 47.9 |
| 8 | 3.30 (dd, 10.3, 5.5) | 79.5 | 4.46 (dd, 11.2, 5.0) | 80.4 |
| 9 | 2.62 (2H, m) | 45.0 | 2.67 (2H, m) | 45.0 |
| 10 | 2.26 (m) | 26.2 | 2.25 (m) | 26.4 |
| 2.03 (m) | | 2.01 (m) | |
| 11 | 5.40 (t, 4.1) | 118.8 | 5.49 (t, 4.4) | 120.2 |
| 12 | | 139.5 | | 138.3 |
| 13 | | 56.0 | | 55.7 |
| 14 | 2.21 (dd, 13.1, 6.2) | 31.6 | 2.25 (m) | 31.8 |
| 1.05 (m) | | 1.10 (t, 12.4) | |
| 15 | 2.72 (m) | 31.6 | 2.94 (m) | 28.6 |
| 16 | 1.00 (3H, d, 6.2) | 20.3 | 0.88 (3H, d, 6.2) | 19.8 |
| 17 | | 172.7 | | 172.2 |
| 18 | 2.58 (2H, t, 6.9) | 36.4 | 2.57 (2H, t, 8.1) | 36.3 |
| 19 | 2.85 (2H, t, 6.9) | 30.7 | 2.85 (2H, td, 7.5, 2.1) | 30.7 |
| 20 | | 132.1 | | 132.1 |
| 21 | 6.68 (d, 2.1) | 110.9 | 6.68 (d, 1.8) | 110.8 |
| 22 | | 146.4 | | 146.4 |
| 23 | | 144.0 | | 144.0 |
| 24 | 6.82 (d, 8.2) | 114.3 | 6.82 (d, 7.9) | 114.3 |
| 25 | 6.68 (dd, 8.2, 2.1) | 120.8 | 6.66 (dd, 7.9, 1.8) | 120.8 |
| 26 | 3.88 (3H, s) | 55.9 | 3.88 (3H, s) | 55.9 |
| COMe | | | | 170.9 |
| COMe | | | 2.12 (3H, s) | 21.3 |
a) Measured at 600 and 150 MHz, respectively.
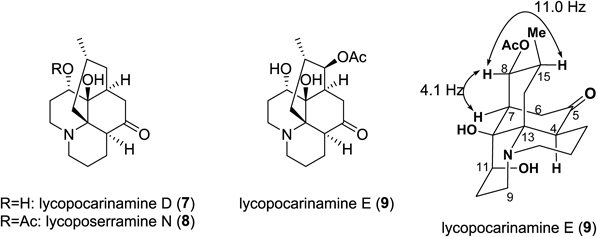
New alkaloid 7, named lycopocarinamine D, was obtained as a colorless amorphous powder and its molecular formula was established as C16H25NO3 by HR-ESI-MS [m/z 280.18909 (MH)+] (Fig. 4). 1H-NMR data (Table 3) showed signals characteristic of common Lycopodium alkaloids, namely, one secondary methyl group [δ 0.87 (3H, d, H3-16)] and two sets of aminomethylenes [δ 3.22 and δ 2.50 (H2-1), δ 3.53 and δ 2.45 (H2-9)]. 13C-NMR data indicated the presence of a ketone carbon [δ 214.8 (C-5)], an oxymethine carbon [δ 74.3 (C-11)], and two quaternary carbons [δ 70.0 (C-12), δ 60.9 (C-13)] attached to the heteroatom. The 1H- and 13C-NMR data were similar to those of known alkaloid lycoposerramine N (8)26) except for the absence of an acetyl group in 7. From the data mentioned above, 7 was deduced to be a deacetyl derivative of lycoposerramine N (8).
Table 3. NMR Data for
7–
9 in CDCl
3a)| Position | Lycopocarinamine D (7) | Lycopocarinamine E (9) | Lycoposerramine N (8) |
|---|
| 1H | 13C | 1H | 13C | 1H | 13C |
|---|
| 1 | 3.22 (td, 13.8, 3.4) | 46.6 | 3.16 (td, 14.4, 4.1) | 47.0 | 3.22 (td, 14.2, 3.8) | 46.6 |
| 2.50 (overlapped) | | 2.54 (overlapped) | | 2.49 (dd, 14.7, 5.2) | |
| 2 | 1.90 (m) | 18.0 | 1.90 (m) | 17.8 | 1.90 (qt, 13.7, 5.4) | 18.0 |
| 1.37 (br d, 13.8) | | 1.38 (overlapped) | | 1.40 (br d, 15.0) | |
| 3 | 2.05 (m) | 19.4 | 2.04 (br d, 11.0) | 19.0 | 2.10 (br d, 12.5) | 19.7 |
| 1.67–1.57 (overlapped) | | 1.69–1.50 (overlapped) | | 1.66 (m) | |
| 4 | 3.72 (m) | 42.5 | 3.76 (dd, 12.4, 4.1) | 42.4 | 3.44 (dd, 11.9, 3.7) | 42.4 |
| 5 | | 214.8 | | 213.7 | | 213.4 |
| 6 | 3.48 (dd, 16.5, 6.9) | 45.6 | 3.21 (dd, 17.2, 6.9) | 42.5 | 2.85 (dd, 17.2, 6.9) | 45.2 |
| 2.28 (d, 16.5) | | 2.44–2.40 (overlapped) | | 2.32 (dd, 17.4, 1.2) | |
| 7 | 2.26 (overlapped) | 39.7 | 2.44–2.40 (overlapped) | 38.6 | 2.20 (t, 3.5) | 38.9 |
| 8 | 1.96 (td, 13.1, 3.4) | 37.3 | 5.24 (dd, 11.0, 4.1) | 74.1 | 2.00 (td, 13.1, 4.0) | 37.3 |
| 1.32 (br d, 11.0) | | | | 1.31 (br d, 11.9) | |
| 9 | 3.53 (br t, 13.1) | 41.4 | 3.51 (td, 14.4, 5.5) | 41.3 | 3.33 (td, 12.8, 3.1) | 41.9 |
| 2.45 (m) | | 2.57–2.51 (overlapped) | | 2.46 (m) | |
| 10 | 2.50 (overlapped) | 31.4 | 2.57–2.51 (overlapped) | 31.9 | 2.42 (m) | 27.1 |
| 1.67–1.57 (overlapped) | | 1.69–1.50 (overlapped) | | 1.75 (dq, 15.0, 2.5) | |
| 11 | 4.01 (s) | 74.3 | 4.05 (d. 1.4) | 71.5 | 4.98 (t, 2.7) | 75.2 |
| 12 | | 70.0 | | 64.3 | | 69.7 |
| 13 | | 60.9 | | 61.0 | | 60.7 |
| 14 | 2.34 (dd, 13.8, 4.8) | 37.0 | 2.44–2.40 (overlapped) | 35.8 | 2.38 (m) | 37.1 |
| 1.26 (overlapped) | | 1.40–1.36 (overlapped) | | 1.26 (t, 13.1) | |
| 15 | 1.51 (m) | 24.1 | 1.69–1.50 (overlapped) | 30.9 | 1.48 (m) | 24.1 |
| 16 | 0.87 (3H, d, 6.2) | 22.4 | 0.88 (3H, d, 6.2) | 22.7 | 0.87 (3H, d, 6.1) | 22.4 |
| COMe | | | | 170.6 | | 169.5 |
| COMe | | | 2.05 (3H, s) | 21.2 | 2.07 (3H, s) | 21.9 |
a) Measured at 600 and 150 MHz, respectively.
The molecular formula of new alkaloid 9, named lycopocarinamine E, was deduced to be C18H27NO5 from HR-ESI-MS [m/z 338.19654 (MH)+]. The 1H- and 13C-NMR data markedly resembled those of lycopocarinamine D (7) except for the chemical shifts of H-8 and C-8 and the existence of an acetoxy group [δΗ 2.05 (3H, s), and δC 170.6 and δC 21.2] in 9. Judging from these data, 9 was deduced to be an 8-acetoxy derivative of 7. The configuration of the acetoxy group at C-8 was shown to be β by the coupling constant between H-8 and H-15 (J8,15=11.0 Hz). Lycopocarinamine E (9) is the first example of lycopodine-type alkaloids having three hydroxy groups at C-8, C-11, and C-12 positions.
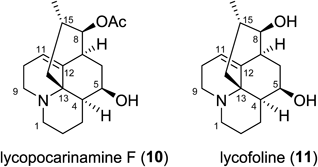
The 1H-NMR spectrum of lycopocarinamine F (10) showed specific signals due to Lycopodium alkaloids, namely, one secondary methyl group [δ 0.89 (3H, d, H3-16)], two sets of aminomethylenes [δ 2.98 and δ 2.55 (H2-1), δ 2.67 (2H, H2-9)], and an olefinic proton [δ 5.53 (H-11)] (Table 4). The 1H- and 13C-NMR spectra evidently resembled those of known alkaloid lycofoline (11)31) except for the existence of signals for an acetyl group [δH 2.09 (3H, s), δC 171.0 and δC 21.4] in 10. The molecular formula of 10 was established by HR-ESI-MS [m/z 306.20928 (MH)+] as C18H27NO3, which indicated that 10 had an extra C2H2O compared to lycofoline (11). From the data mentioned above, 10 must be an acetyl derivative of lycofoline (11). The chemical shift of H-8 in 10 was lower than that in lycofoline (11), indicating that the acetyl group was attached to O-8 position (Fig. 5).
Table 4. NMR Data for
10 and
11 in CDCl
3a)| Position | Lycopocarinamine F (10) | Lycofoline (11) |
|---|
| 1H | 13C | 1H | 13C |
|---|
| 1 | 2.98 (t-like, 9.6) | 48.5 | 2.96 (m) | 48.6 |
| 2.55 (d, 11.0) | | 2.54 (m) | |
| 2 | 1.60 (2H, m) | 24.7 | 1.77 (m) | 24.9 |
| | | 1.62 (m) | |
| 3 | 1.78 (m) | 23.2 | 1.77 (m) | 23.1 |
| 1.51 (m) | | 1.48 (m) | |
| 4 | 2.05–1.88 (overlapped) | 47.1 | 1.94 (m) | 47.6 |
| 5 | 3.97 (t, 4.8) | 67.8 | 3.96 (t, 5.5) | 68.0 |
| 6 | 2.05–1.88 (2H, overlapped) | 33.0 | 2.12 (d, 15.8) | 32.6 |
| | | 1.89 (dt, 15.8, 6.2) | |
| 7 | 2.70 (t, 4.8) | 42.5 | 2.54 (m) | 45.8 |
| 8 | 4.44 (dd, 10.2, 4.8) | 81.5 | 3.23 (br s) | 79.9 |
| 9 | 2.67 (2H, m) | 45.1 | 2.66 (2H, m) | 45.1 |
| 10 | 2.22 (m) | 26.3 | 2.24 (m) | 26.2 |
| 2.05–1.88 (overlapped) | | 2.01 (m) | |
| 11 | 5.53 (t, 4.1) | 117.9 | 5.45 (t, 3.7) | 116.4 |
| 12 | | 142.1 | | 143.5 |
| 13 | | 56.1 | | 56.4 |
| 14 | 2.28 (dd, 13.7, 6.8) | 32.5 | 2.22 (dd, 13.0, 6.8) | 32.4 |
| 1.10 (t, 13.0) | | 1.06 (t, 13.0) | |
| 15 | 3.07 (m) | 28.1 | 2.77 (m) | 31.5 |
| 16 | 0.89 (3H, d, 6.2) | 19.8 | 1.02 (3H, d, 6.9) | 20.4 |
| COMe | | 171.0 | | |
| COMe | 2.09 (3H, s) | 21.4 | | |
a) Measured at 600 and 150 MHz, respectively.
Experimental
General1H- and 13C-NMR spectra: JEOL JNM ECA-600 or JEOL ECZ-600 or ECP-600 at 600 MHz (1H-NMR) and 150 MHz (13C-NMR), respectively. HR-ESI-MS: JEOL AccuTOF-GCv. UV: JASCO V-560. Optical rotation: JASCO P-1020. TLC: precoated silica gel 60 F254 plates (Merck, 0.25 mm thick), NH-TLC (Fuji Silysia Chemical). Flash column chromatography: Silica gel 60N (Kanto Chemical, 40–50 µm). Amino-silica gel column chromatography: NH-DM1020 (Fuji Silysia Chemical).
Plant MaterialLycopodium carinatum was purchased from a plant market in Bangkok. The plant was identified by Dr. S. Wongseripipatana and a voucher specimen (No.20100201) is stored at the Graduate School of Pharmaceutical Sciences, Chiba University, Japan.
Extraction and IsolationThe dried, powdered whole plant (451.2 g) was extracted with MeOH four times (twice at room temperature, twice under reflux). The extract was evaporated to give a crude MeOH extract (150.2 g). The MeOH extract was dissolved in 2% aq. tartaric acid (2.0 L) and the mixture was filtered to remove the insoluble part. The filtrate was extracted with petroleum ether (1.0 L) and the acidic aq. layer was basified with Na2CO3 (pH 10) and extracted with 5% MeOH/CHCl3 (1.75 L×4). The organic layer was dried over MgSO4, filtered, and evaporated to give the crude alkaloidal fraction (2.99 g).
The crude alkaloidal fraction was separated by SiO2 flash chromatography using MeOH/CHCl3 gradient (3–50%), MeOH, and 10% aq. ammonia/MeOH to give 13 fractions (frs. A–M). The MeOH eluate (fr. J) was purified by amino-silica gel open column chromatography with 5% MeOH/AcOEt, MeOH, and 10% aq. ammonia/MeOH to give six fractions (frs. J1–J6). The 5% MeOH/AcOEt eluate (fr. J4) was purified successively by amino-silica gel open column chromatography (10–20% EtOH/n-hexane, 2% MeOH/CHCl3) to afford lycopocarinamine A (4.2 mg), lycopocarinamine B (1.7 mg), lycopocarinamine C (1.8 mg), lycopocarinamine D (0.9 mg), and lycopocarinamine E (0.3 mg). Lycopocarinamine F (3.3 mg) was isolated from fr. J2 by SiO2 flash column chromatography (3% MeOH/CHCl3 saturated with ammonia).
Other isolated compounds from the crude alkaloidal fraction were lycopodine,31) lycocarinatine A,28) 12-epi-lycodoline N-oxide,32) lycofoline,30) anhydrolycodoline,30) obscurumine C,33) gnidioidine,30) malycorin B,29) fawcettimine,34) lycoposerramine-C,35) lycoposerramine-D,35) lycoposerramine-P,35) phlegmariurine A,22) phlegmariurine B,36) huperzine A,5) huperzine C,37) 13N-formyl huperzine A,38) lycodoline,32,39) and lycodine.18)
Lycopocarinamine A (1)Colorless amorphous powder; 1H- and 13C-NMR, see Table 1; [α]D27 −8.9 (c=0.14, MeOH); UV λmax (MeOH) 282.5, 229.5, 216.0 nm; HR-ESI-MS m/z 460.26722 (MH)+ (Calcd for C26H38NO6: 460.26991)
Lycopocarinamine B (2)Colorless amorphous powder; 1H- and 13C-NMR, see Table 1; [α]D27 −10.8 (c=0.11, MeOH); UV λmax (MeOH) 279.5, 229.5, 206.0 nm; HR-ESI-MS m/z 474.28222 (MH)+ (Calcd for C27H40NO6: 474.28556)
Lycopocarinamine C (5)Colorless amorphous powder; 1H- and 13C-NMR, see Table 2; [α]D26 −4.0 (c=0.08, MeOH); UV λmax (MeOH) 281.5, 230.0, 215.5 nm; HR-ESI-MS m/z 458.25452 (MH)+ (Calcd for C26H36NO6: 458.25426)
Lycopocarinamine D (7)Colorless amorphous powder; 1H- and 13C-NMR, see Table 3; [α]D24 −9.5 (c=0.04, MeOH); HR-ESI-MS m/z 280.18909 (MH)+ (Calcd for C16H26NO3: 280.19127)
Lycopocarinamine E (9)Colorless amorphous powder; 1H- and 13C-NMR, see Table 3; [α]D25 +44.8 (c=0.008, MeOH); HR-ESI-MS m/z 338.19654 (MH)+ (Calcd for C18H28NO5: 338.19675)
Lycopocarinamine F (10)Colorless amorphous powder; 1H- and 13C-NMR, see Table 4; [α]D25 −3.4 (c=0.14, MeOH); HR-ESI-MS m/z 306.20928 (MH)+ (Calcd for C18H28NO3: 306.20692)
Chemical Conversion of Lycopocarinamine A (1) into Lycopocarinamine B (2)To a solution of lycopocarinamine A (1, 1.0 mg) in MeOH (0.5 mL) was added freshly prepared ether solution of CH2N2 (excess). The mixture was stirred for 1 h at room temperature. After the decomposition of CH2N2 (the yellowish color faded) was completed, the solvent was removed under reduced pressure. The residue was purified by SiO2 flash chromatography (MeOH/CHCl3 gradient) to give semi-synthetic 2 (0.37 mg, 36%). All the spectroscopic data of semi-synthetic 2 were identical with those of natural 2.
Chemical Conversion of Lycopocarinamine A (1) into Lycocarinatine A (3)To a stirred solution of 1 (0.5 mg) in dry CH2Cl2 (0.1 mL) was added mCPBA (77%, 0.27 mg) at 0°C under argon atmosphere. After the reaction mixture was stirred at 0°C for 30 min, it was directly subjected to amino-silica gel open column chromatography (MeOH/AcOEt gradient) to give semi-synthetic 3 (0.67 mg, quant.). All the spectroscopic data of semi-synthetic 3 were identical with those of natural 3.