Regular Articles
Introducing Telescoping Process to Synthesis of a Key Intermediate of Drug Discoveries Using Design of Experiment
2016 Volume 64 Issue 7 Pages 1043-1046

Details
2016 Volume 64 Issue 7 Pages 1043-1046
The 5-bromo-2-methylamino-8-methoxyquinazoline (1) is a key intermediate in our drug discoveries. Compound 1 bears a monomethylamino group at the 2-position of the quinazoline ring. This compound has been synthesized from 6-bromo-2-fluoro-3-methoxybenzaldehyde by a synthetic route including a total of four isolation processes in the medicinal chemistry laboratories. Our process chemistry laboratories successfully improved the original synthetic route by introducing the telescoping process. We successfully reduced the isolation processes from four to two processes by using information extracted through design of experiment. The total yield of compound 1 increased by 18%, while maintaining the purity of compound 1 of the original synthetic route. Accordingly, we contributed to the quick supply of compound 1 to the medicinal laboratories.
The 5-bromo-2-methylamino-8-methoxyquinazoline (1) is a key intermediate in our drug discoveries. In the medicinal chemistry laboratories, various drug derivatives have been synthesized from compound 1 via the Suzuki–Miyaura coupling reaction at the 5-bromo position by using various boronic acids. Our process chemistry laboratories played an important role to supply the key intermediate—compound 1—quickly to the medicinal chemistry laboratories, in order to find the drug candidate as soon as possible. Compound 1 is a compound bears a monomethylamino group at the 2-position of the quinazoline ring. In the medicinal chemistry laboratories, this compound has originally been synthesized by the synthetic route via monomethylation after protection of the 2-amino group by the acetyl group to avoid dimethylation of the 2-amino group (Chart 1).

That is, compound 1 was synthesized from 6-bromo-2-fluoro-3-methoxybenzaldehyde (2) as follows: the quinazoline ring formation using guanidine at a high temperature (1st step), protection of the 2-amino group of the quinazoline ring using acetic anhydride (2nd step), methylation for the 2-acetylamino group (3rd step) and deprotection of the acetyl group using methanol (4th step). Furthermore, each intermediate (compounds 3–5) was isolated as a crystal solid, and accordingly the original synthetic route was lengthy. On the other hand, an alternative shorter route via the direct quinazoline ring formation from compound 2 using N-methyl guanidine was attractive; however this attempt didn’t succeed. The complex mixture was generated, and no trace of quinazoline compounds was detected. After all, we thought that the original synthetic route was realistic although it was lengthy. If the telescoping process1) was introduced to the original synthetic route by unified solvents between consecutive reaction steps and avoiding some isolation processes of intermediates, the original synthetic route might be transformed to the simple and efficient synthetic route. We tried to improve the original synthetic route with the aim to introduce the telescoping process. We also used design of experiment (DoE)2) when optimizing some reaction steps. Results of our investigation were reported below.
A high temperature was needed for the quinazoline ring formation from compound 2 and guanidine; therefore N,N-dimethylacetamide with a high boiling point has been used as a solvent. On the other hand, in acetylation, a large amount of pyridine which was a malodorous and poisonous solvent was used as a solvent as well as a base. If N,N-dimethylacetamide of the solvent of the 1st step was useable as an alternative solvent for acetylation, it might not only reduce the amount of pyridine but also introduce the telescoping process between the 1st and 2nd steps. We noticed an interesting result for pyridine, when we studied to optimize the reaction condition of acetylation by DoE. In that experiment, four parameters (A: amount of acetic anhydride, B: amount of pyridine, C: amount of N,N-dimethylacetamide, and D: temperature) were chosen as optimizing parameters in the 2-level fractional factorial design (Table 1).
| Level | Parametera) | |||
|---|---|---|---|---|
| A (eq) | B (eq) | C (v/w) | D (°C) | |
| Low (−) | 1.0 | 2.0 | 10 | 60 |
| High (+) | 3.0 | 6.0 | 30 | 100 |
a) A: amount of acetic anhydride. B: Amount of pyridine. C: Amount of N,N-dimethylacetamide. D: Temperature.
This experimental plan and result of the yield of compound 4 are shown in Table 2.
| Run | Parameterb) | Yield of compound 4b) (%) | |||
|---|---|---|---|---|---|
| A | B | C | D | ||
| 1 | − | − | − | − | 15 |
| 2 | + | − | − | + | 95 |
| 3 | − | + | − | + | 61 |
| 4 | + | + | − | − | 39 |
| 5 | − | − | + | + | 47 |
| 6 | + | − | + | − | 23 |
| 7 | − | + | + | − | 8 |
| 8 | + | + | + | + | 86 |
a) All reactions were carried out for 4 h on a 100 mg scale. b) Product ratios were calculated by HPLC assay.
The results of experimental runs of the 2-level fractional factorial design were analyzed by the software Design Expert®. The half normal plot outputted from the software is shown in Fig. 1.
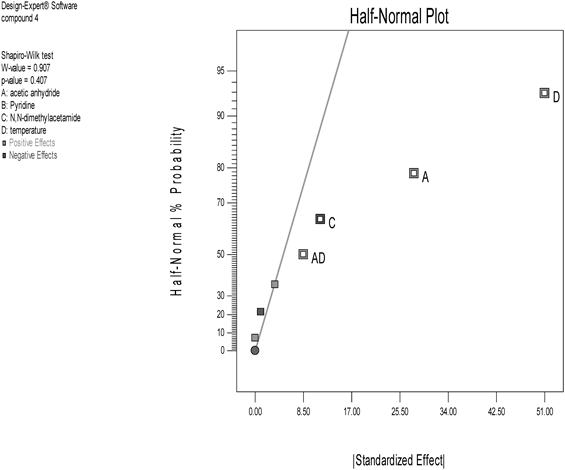
Total four points (A, C, D, and AD which is interaction between A and D) diverged from the line to the right. This result indicates that three parameters, A, C and D, were extracted as effective parameters for acetylation. Surprisingly, parameter B—pyridine—was not extracted, although we thought that a base generally played an important role for acetylation. This interesting result motivated us to investigate this acetylation without using pyridine. We actually experimented acetylation of compound 3 using N,N-dimethylacetamide in absence of pyridine. In this experiment, the amount of N,N-dimethylacetamide was set on the low level, and the amount of acetic anhydride and the temperature were set on the high level by referring to the cube graph obtained from the DoE investigation (Fig. 2).

As a result, we confirmed that acetylation proceeded as expected3); furthermore the isolated yield of compound 4 was equivalent to the case using pyridine (Table 3).
| Run | Pyridine (eq) | Isolated yield of compound 4 (%) |
|---|---|---|
| 1b) | 2.0 | 91 |
| 2c) | Not added | 95 |
a) All reactions were carried out using 3.0 eq of acetic anhydride and 10 v/w of N,N-dimethylacetamide at 100°C for 4 h. b) Run 1 was carried out on the 2.6 g scale. c) Run 2 was carried out on the 1.0 g scale.
From the above results, we tried to investigate the telescoping process between the 1st and 2nd steps by using N,N-dimethylacetamide as a common solvent. Compound 2 was dissolved in N,N-dimethylacetamide (11 v/w), and the quinazoline ring formation was completed by mixing with guanidine carbonate (3.0 eq) at 130°C for a 0.5 h. The reaction mixture including compound 3 was filtrated at 60°C. An insoluble residual on the filter (hydrogen fluoride salt of guanidine) was washed by N,N-dimethylacetamide (3.7 v/w) and removed. Then acetic anhydride (3.0 eq) was added to the filtrate solution, and compound 4 was isolated in 89% yield after acetylation at 100°C for 4 h. We successfully obtained compound 4 directly from compound 2 without isolating compound 3. In this telescoping process, the total amount of N,N-dimethylacetamide was set on the 14.7 v/w because the additional amount of N,N-dimethylacetamide was needed for the filtration process of an insoluble residual as mentioned above.
Introducing the Telescoping Process between the 3rd and 4th StepsWe also found the interesting result when optimizing methylation using iodomethane by DoE. Four parameters (A: amount of potassium carbonate, B: amount of iodomethane, C: temperature, and D: amount of N,N-dimethylformamide) were chosen (Table 4), and the yield of compound 5 was optimized by the 2-level fractional factorial design (Table 5).
| Level | Parametera) | |||
|---|---|---|---|---|
| A (eq) | B (eq) | C (°C)b) | D (v/w) | |
| Low (−) | 1.0 | 1.0 | 20 | 10 |
| High (+) | 3.0 | 3.0 | 40 | 30 |
a) A: Amount of potassium carbonate. B: Amount of iodomethane. C: Temperature. D: Amount of N,N-dimethylformamide. b) The level of temperature was set under a boiling point of iodomethane (42°C).
| Run | Parameterb) | Yield of compound 5b) (%) | |||
|---|---|---|---|---|---|
| A | B | C | D | ||
| 1 | − | − | − | − | 36 |
| 2 | + | − | − | + | 90 |
| 3 | − | + | − | + | 37 |
| 4 | + | + | − | − | 91 |
| 5 | − | − | + | + | 39 |
| 6 | + | − | + | − | 97 |
| 7 | − | + | + | − | 35 |
| 8 | + | + | + | + | 92 |
a) All reactions were carried out for 4 h on a 100 mg scale. b) Product ratios were calculated by HPLC assay.
As a result, the very simple result was obtained in the half-normal plot (Fig. 3).

The parameter A—potassium carbonate—was only extracted as an effective parameter. This result indicates that we should only take care of the amount of potassium carbonate for this methylation and can freely set each value of the other three parameters within the investigated experimental space of DoE. On the other hand, potassium carbonate was also used as a base for deacetylation of compound 5 in the original synthetic route. If methanol used as a nucleophile of deacetylation was added to the reaction mixture after completion of methylation, deacetylation might proceed on the telescoping process. We actually tried to investigate the telescoping process between the 3rd and 4th steps. Here the amount of potassium carbonate was set on the high level (3.0 eq) by referring to the one factor graph obtained from the DoE investigation of methylation (Fig. 4).

Each amount of iodomethane and N,N-dimethylformamide was set on the low level in consideration of operability of the telescoping process. The temperature was set on 35°C by considering a boiling point of iodomethane, and this value can easily be controlled by the water bath in the laboratory when room temperature was controlled under 30°C. Compound 4 was dissolved in N,N-dimethylformamide (10 v/w). Then potassium carbonate (3.0 eq) and iodomethane (1.1 eq) were added to this solution, and methylation was completed at 35°C for 4 h. After methanol (5 v/w) was added to the reaction mixture, deacetylation finished at 60°C for 1 h without adding an extra amount of potassium carbonate. We successfully obtained compound 1 directly from compound 4 in the 94% yield without isolating compound 5.
The original synthetic route of compound 1 was improved to the efficient synthetic route by introducing the telescoping process (Chart 2). In the improved synthetic route, compound 1 was successfully synthesized via two isolation processes. The total yield of the compound 1 from compound 2 increased by 18%, with the purity of compound 1 equivalent to that obtained by the original synthetic route (the original: 98.6%, the improved: 98.8% by HPLC area). This report also demonstrates that the results obtained from DoE provided useful information for introducing the telescoping process. Our process chemistry laboratories successfully established this improved process using DoE, and contributed to the quick supply of compound 1 to the medicinal laboratories.

All reagents and solvents are commercially available (ex. Sigma-Aldrich Co.) and used without further purification. Melting point was obtained on a Mettler FP61. 1H-NMR spectra were obtained on a JEOL JNM-LA300 spectrometer (300 MHz). Chemical shifts (δ) are reported in ppm. Multiplicities are given as: s (singlet), d (doublet), br s (broad singlet). Mass spectra was obtained on a Micromass LCT spectrometer. HPLC analyses were performed on a Hitachi L-7000 system.
HPLC AnalysesThe HPLC data in Tables 3 and 5 were obtained under the following conditions: detector, ultraviolet absorption photometer (wavelength 254 nm); column, Imtakt Cadenza CD-C18; mobile phase, a mixture of 0.01 mol/L KH2PO4 and 0.01 mol/L K2HPO4–CH3CN (2 : 5); flow rate, 1.0 mL/min; column temperature, 35°C; retension time (tR) (min) compound 4 (4.0), compound 1 (10.0).
Synthesis of Compound 4Compound 2 (3.0 g) was added to a suspension of guanidine carbonate (3.5 g, 3.0 eq) in N,N-dimethylacetamide (33 mL, 11 v/w). The mixture was stirred at 130°C for a 0.5 h, and filtrated at 60°C. An insoluble residual on the filter was washed by N,N-dimethylacetamide (11 mL, 3.7 v/w), and then acetic anhydride (3.6 mL, 3.0 eq) was added to the filtrate solution. The mixture was stirred at 100°C for 4 h, and then stirred at 0°C for 1 h. The solid was corrected by filtration to give compound 4 (3.4 g, 89%) as a white colorless crystal; mp 267–268°C; 1H-NMR (DMSO-d6, 300 MHz) δ: 10.9 (br s, 1H), 9.46 (s, 1H), 7.77 (d, J=8.4 Hz, 1H), 7.32 (d, J=8.4 Hz, 1H), 3.96 (s, 3H), 2.29 (s, 3H); MS electrospray ionization (ESI) (+) m/z 296 (79Br), 298 (81Br) [M+H]+.
Synthesis of Compound 1Compound 4 (2.0 g) was added to a mixture of potassium carbonate (2.8 g, 3.0 eq), iodomethane (0.46 mL, 1.1 eq) in N,N-dimethylformamide (20 mL, 10 v/w). The mixture was stirred at 35°C for 4 h, and then methanol (10 mL, 5 v/w) was added. The mixture was stirred at 60°C for 1 h, and then water (20 mL, 10 v/w) was added. The suspension stirred at 0°C for 1 h. The solid was corrected by filtration to give compound 1 (1.8 g, 94%) as a yellow crystal; mp 241–242°C; 1H-NMR (DMSO-d6, 300 MHz) δ: 9.26 (s, 1H), 7.29 (d, J=8.4 Hz, 1H), 6.89 (d, J=8.4 Hz, 1H), 5.46 (br s, 1H), 4.00 (s, 3H), 3.17 (s, J=5.1 Hz, 3H); MS ESI (+) m/z 268 (79Br), 270 (81Br) [M+H]+.
The authors declare no conflict of interest.