Special Collection of Papers: Regular Articles
Convergent Synthesis of 2-Aryl-Substituted Quinolines by Gold-Catalyzed Cascade Reaction
2016 Volume 64 Issue 7 Pages 824-829
Details
2016 Volume 64 Issue 7 Pages 824-829
Gold-catalyzed auto-tandem catalysis has been developed for synthesizing 2-aryl-substituted quinolines. The reaction of an aniline bearing an acetal moiety with an aryl alkyne proceeded via formal [4+2]-cycloaddition, which involved the addition of gold acetylide to an oxonium ion to give amino alkyne intermediate and sequential 6-endo-dig cyclization of amino alkyne intermediate by attacking of nitrogen to alkyne moiety activated by gold catalyst. The cationic gold catalyst promoted two different processes by enhancing the nucleophilicity and electrophilicity of alkyne. This convergent synthetic methodology enabled the synthesis of a variety of 2-aryl-substituted quinolines.
A quinoline skeleton is often observed in biologically important natural products, for example; quinoline alkaloid quinine exhibits medicinally vital antimalarial activity.1–4) Among quinoline compounds, 2-arylquinoline has recently attracted attention from biologists due to its unique biological activity.5–7) Therefore, quinoline compounds have inspired the development of new synthetic methods8,9) for the formation of the N-heterocyclic scaffold due to their potential utility in the pharmaceutical industry. As classical and conventional synthetic methods, the Skraup,10) Doebner and von Miller,11) Combes,12) Friedländer,13) and Pfitzinger14) quinoline syntheses are well known; however, these methods often encounter problems with functional group compatibility as they require strong acidic or basic conditions. In recent decades, transition metal-catalyzed syntheses of substituted quinolines with a highly convergent manner using an alkyne have also been developed.15–22) These reactions proceed under relatively mild conditions and enable the synthesis of various multi-substituted quinolines.
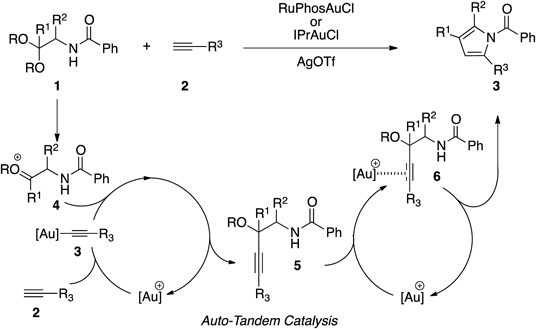
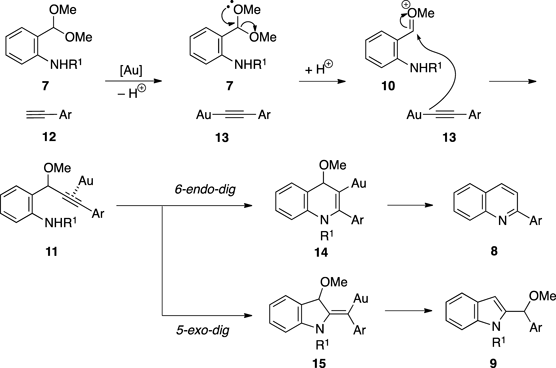
Recently, we developed a novel gold-catalyzed multi-substituted pyrrole synthesis.23,24) This reaction is highly versatile, involving the assembly of two acyclic substrates 1 and 2 with substituents installed beforehand. In addition, a cascade process involving gold-mediated auto-tandem catalysis increases the efficiency and convergence of the reaction. On the basis of the advantages of this reaction, we applied this gold-catalyzed cascade reaction to syntheses of other N-heterocyclic compounds. Considering the above information, we envisioned that the corresponding reaction of aniline 7 bearing an acetal moiety at the ortho-position with alkyne 12 should furnish a quinoline 8 or an indole derivative 9. Thus, an intermolecular addition of gold acetylide to the oxonium ion 10 should produce propargyl ether 11. Then, cyclization via intramolecular nucleophilic addition of nitrogen to the alkyne activated by the gold catalyst would occur in a 6-endo-dig fashion and, following aromatization by removal of the protecting group, would afford quinoline derivative 8. On the contrary, the 5-exo-dig cyclization of 11 would give indole derivative 9.
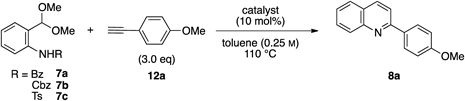
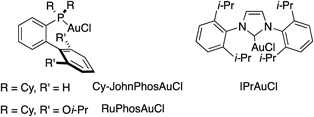
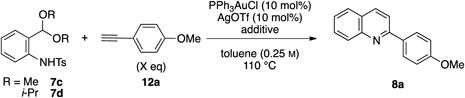
At the outset, we examined the reaction of anilide 7a with a benzoyl (Bz) group and 4-methoxyphenylacetylene 12a (Table 1). Treatment of 7a and 12a in the presence of 10 mol% PPh3AuCl and AgOTf (Tf, trifluoromethanesulfonyl) in toluene solution at 110°C gave a quinoline 8a in 7% yield without the formation of an indole derivative (entry 1). Replacement of the Bz group with a benzyloxycarbonyl (Cbz) group improved the yield of 8a to 20% (entry 2). Anilide 7c with a tosyl (Ts) group gave the desired quinoline 8a in 34% yield (entry 3). We then investigated the ligands of the gold catalyst. Using both trialkylphosphine gold chloride/AgOTf and PPh3AuNTf225) slightly reduced the yield of 8a (entries 4, 5). Next, a series of catalysts26,27) with bulky biphenyl moieties (entries 6, 7) and an N-heterocyclic carbene gold catalyst IPrAuCl28) (entry 8) were examined; however, these produced no improvement in the yield of 8a. It was found that independently using AgOTf or PPh3AuCl substantially prolonged the reaction time (entries 9, 10). Finally, we conducted the reaction using CuI based on the known quinoline synthesis,19) but the desired quinoline 8a was obtained in only trace amounts (entry 11).
 | ||||
|---|---|---|---|---|
| Entry | R | Catalyst | Time (h) | Yield (%) |
| 1 | Bz | Ph3PAuCl/AgOTf | 2 | 7 |
| 2 | Cbz | Ph3PAuCl/AgOTf | 2 | 20 |
| 3 | Ts | Ph3PAuCl/AgOTf | 2 | 34 |
| 4 | Ts | Et3PAuCl/AgOTf | 3 | 29 |
| 5 | Ts | Ph3PAuNTf2 | 3 | 29 |
| 6 | Ts | RuPhosAuCl/AgOTf | 1 | 31 |
| 7 | Ts | Cy-JohnPhosAuCl/AgOTf | 1 | 25 |
| 8 | Ts | IPrAuCl/AgOTf | 1 | 26 |
| 9 | Ts | Ph3PAuCl | 28 | 5 |
| 10 | Ts | AgOTf | 15 | 26 |
| 11 | Ts | CuI | 13 | trace |
To improve the yield, we investigated reaction conditions of 7c and 12a with various additives (Table 2). As described in our previous paper,29) we initially added KHSO4 as a proton source to promote the catalytic cycle by accelerating protodeauration step (entry 1). Although the yield of quinoline 8a was increased, the decomposition of the acetal moiety of 7c was facilitated to provide the aldehyde 16 in 57% yield. Thus, we envisaged that addition of MeOH would suppress the decomposition of the acetal moiety by inducing an oxonium-acetal equilibrium. As expected, aldehyde generation was suppressed by addition of MeOH; however, the yield of quinoline was remained constant (entry 2). We thought this reason would be attributed to consumption of alkyne 12a via gold-catalyzed methanolysis. Increasing amount of alkyne 12a to 5 eq improved the yield of 8a to 49% yield (entry 3). After extensive optimizations, we established the optimal conditions using sterically demanding alcohol, such as i-PrOH, with 2,6-di-t-butylpyridine (DTBP) to afford the target quinoline in 66% yield (entry 6). Treatment of diisopropyl acetal 7d instead of dimethyl acetal 7c with optimal conditions led to a decreased yield of 8a to 45% (entry 7). Finally, when we conducted this reaction on sub-gram scale (1.55 mmol), the reaction proceeded smoothly to give the desired quinolone 8a in 62% yield (entry 8).
 | ||||||
|---|---|---|---|---|---|---|
| Entry | R | X (equiv) | Additive | Time (h) | Yield (%) | |
| 1a) | Me | 3 | KHSO4 (15 mol%) | 2 | 39 | 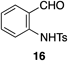 |
| 2b) | Me | 3 | MeOH (5 equiv) | 2 | 38 | |
| 3 | Me | 5 | MeOH (5 equiv) | 2 | 49 | |
| 4 | Me | 5 | MeOH (20 equiv) | 2 | 50 | |
| 5 | Me | 5 | MeOH (20 equiv) 2,6-di-t-butylpyridine (40 mol%) | 3 | 44 | |
| 6 | Me | 5 | i-PrOH (20 equiv) 2,6-di-t-butylpyridine (40 mol%) | 7 | 66 | |
| 7 | i-Pr | 5 | i-PrOH (20 equiv) 2,6-di-t-butylpyridine (40 mol%) | 4 | 45 | |
| 8c) | Me | 5 | i-PrOH (20 equiv) 2,6-di-t-butylpyridine (40 mol%) | 7 | 62 | |
a) Aldehyde 16 was obtained in 57% yield. b) Aldehyde 16 was obtained in 17% yield. c) The reaction on sub-gram scale (7c: 500 mg).
Having established the optimal conditions, we then focused on the alkyne scope of the reaction (Table 2). Phenylacetylene (12b) and substituted phenyl groups with a methyl and bromo groups at their para position (12c, d) uneventfully gave the corresponding quinoline products in good yields (entries 2–4). The (3,4-dimethoxyphenyl)acetylene 12e also gave the desired product 8e in good yield (entry 5). However, alkynes 12f and g, which bear electron-withdrawing groups, showed lower reactivity and produced decreased yields of the corresponding quinoline 8f and g (entries 6, 7). Additionally, the reaction of 7c with 1-naphthyl alkyne 12h afforded the corresponding quinoline 8h in moderate yield (entry 8).
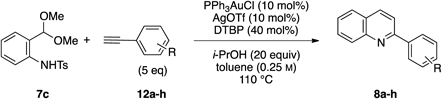 | ||||
|---|---|---|---|---|
| Entry | R | Time (h) | Yield (%) | |
| 1 |  | 8a | 7 | 66 |
| 2 |  | 8b | 7 | 55 |
| 3 | 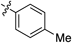 | 8c | 7 | 68 |
| 4 | 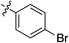 | 8d | 7 | 67 |
| 5 | 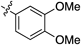 | 8e | 11 | 55 |
| 6 | 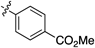 | 8f | 7 | 31 |
| 7 | 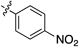 | 8g | 11 | 35 |
| 8 | 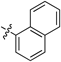 | 8h | 7 | 39 |
In summary, we have developed a new methodology for the synthesis of 2-aryl-substituted quinolines in a cascade manner, which is promoted by auto-tandem gold catalysis. This reaction has the advantages of versatility and convergence toward the synthesis of a variety of 2-arylquinolines, which have recently attracted attention due to its unique biological activity.
Unless otherwise noted, all reactions were performed using oven-dried glassware, sealed with a rubber septum under a slight positive pressure of argon. Anhydrous MeCN and dichloromethane were purchased from Kanto Chemical Co., Inc. Anhydrous i-PrOH, MeOH, toluene, pyridine, and Et3N were dried and distilled according to the standard protocols. Unless otherwise mentioned, materials were obtained from commercial suppliers and used without further purification. Reactions and chromatographical fractions were monitored by TLC analysis with pre-coated silica gel plates 60 F254 (Merck). Flash column chromatography was carried out using Kanto silica gel 60N (spherical, neutral, 40–50 µm). Preparative TLC was performed on pre-coated silica gel plates 60 F254 (Merck). Gel permeation chromatography was carried out using a Japan Analytical Industry Co., Ltd., LC-9201. IR spectra were measured on a Shimadzu FTIR-8300 spectrometer or a JASCO FT/IR-4100 spectrometer. NMR spectra were measured on a JNM-AL400 spectrometer. For 1H spectra, chemical shifts were expressed in ppm downfield from internal tetramethylsilane (δ 0) or relative internal CHCl3 (δ 7.26). For 13C spectra, chemical shifts were expressed in ppm downfield from relative internal CHCl3 (δ 77.0). J-Values were expressed in hertz. Mass spectra were recorded on a Bruker micrOTOF spectrometer.
2-(Dimethoxymethyl)anilineTo a solution of 2-nitrobenzaldehyde (303 mg, 2.00 mmol) and trimethyl orthoformate (0.285 mL, 2.60 mmol) in MeOH (10 mL) was added p-TsOH·H2O (38.0 mg, 0.200 mmol). After stirring for an hour at 70°C, the reaction was quenched with saturated aqueous NaHCO3 and the resulting mixture was extracted with CH2Cl2 three times. The combined organic extracts were washed with brine, dried over Na2SO4, and concentrated under reduced pressure to give the crude acetal. The residue was directly subjected to the next reaction without further purification. A mixture of crude acetal, 10% Pd/C (203 mg, 0.197 mmol), MeOH (2.0 mL) and AcOEt (2.0 mL) was stirred under a hydrogen atmosphere at room temperature for 3 h. The reaction mixture was filtered through a pad of Celite and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (hexanes : AcOEt=9 : 1) to afford aniline (263 mg, 1.58 mmol, 79% over 2 steps) as a yellow oil; Its 1H-NMR spectra data was completely identical with that reported30); 1H-NMR (400 MHz, CDCl3) δ: 7.29 (d, J=7.2 Hz, 1H), 7.13 (dd, J=8.0, 7.2 Hz, 2H), 6.73 (dd, J=8.0, 7.6 Hz, 1H), 6.65 (d, J=7.6 Hz, 1H), 5.32 (s, 1H), 4.23 (br s, 2H), 3.35 (s, 6H).
N-(2-(Dimethoxymethyl)phenyl)benzamide (7a)To a solution of a 2-(dimethoxymethyl)aniline (31.9 mg, 0.191 mmol) in CH2Cl2 (0.65 mL) were added Et3N (0.13 mL, 0.96 mmol) and benzoyl chloride (24 µL, 0.21 mmol) at 0°C. After stirring for an hour, the reaction was quenched with saturated aqueous NH4Cl and the mixture was extracted with AcOEt three times. The combined organic extracts were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by preparative TLC on silica gel (hexanes : AcOEt=8 : 2) to afford benzamide 7a (32.9 mg, 0.121 mmol, 63%) as a pale yellow oil; Rf=0.48 (Silica gel, hexanes : AcOEt=8 : 2); IR attenuated total reflectance (ATR) 3367, 2936, 2830, 1678, 1592, 1533, 1452, 1310, 1066, 759 cm−1; 1H-NMR (400 MHz, CD3CN, 70°C, a mixture of rotamers) δ: 9.61 (br s, 0.8H), 9.38 (br s, 0.2 H), 8.39 (d, J=8.4 Hz, 0.8H), 8.19 (d, J=8.4 Hz, 0.2H), 7.95–7.85 (m, 2H), 7.64–7.49 (m, 3H), 7.45–7.26 (m, 2H), 7.20–7.11 (m, 1H), 5.42 (s, 1H), 3.42 (s, 6H); 13C-NMR (100 MHz, CD3CN, 70°C, a mixture of rotamers) δ: 161.1, 138.3, 136.7, 133.0, 130.6, 130.5, 130.12, 130.07, 129.9, 129.7, 128.5, 128.23, 128.17, 125.4, 124.8, 123.5, 123.0, 106.7, 74.4, 58.6, 55.1; electrospray ionization (ESI)-MS m/z Calcd for C16H17NNaO3 294.1101 (M++Na). Found 294.1104.
Benzyl (2-(Dimethoxymethyl)phenyl)carbamate (7b)To a mixture of the benzyl (2-formylphenyl)carbamate (56.5 mg, 0.221 mmol) and trimethyl orthoformate (36 µL, 0.33 mmol) in MeOH (1.1 mL) was added p-TsOH (3.8 mg, 0.022 mmol). After stirring for 12 h at reflux, the reaction was quenched with saturated aqueous NaHCO3 and the resulting mixture was extracted with CH2Cl2 three times. The combined organic extracts were washed with brine, dried over Na2SO4, and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (hexanes : AcOEt=9 : 1) to afford acetal 7b (49.7 mg, 0.165 mmol, 75%) as a colorless oil; Rf=0.60 (Silica gel, hexanes : AcOEt=8 : 2); IR (neat) 3375, 2938, 1734, 1594, 1531, 1455, 757 cm−1; 1H-NMR (400 MHz, CDCl3) δ: 8.16–8.08 (m, 2H), 7.45–7.32 (m, 7H), 7.03 (dd, J=8.0, 8.0 Hz, 1H), 5.32 (s, 1H), 5.20 (s, 2H), 3.33 (s, 6H); 13C-NMR (100 MHz, CDCl3, a mixture of rotamers) δ: 153.5, 136.3, 132.4, 129.6, 129.3, 128.5, 128.3, 128.2, 128.1, 127.9, 125.2, 124.1, 122.6, 120.2, 103.1, 99.9, 66.8, 54.6, 53.2; ESI-MS m/z Calcd for C17H19NNaO4 324.1212 (M++Na). Found 324.1207.
N-(2-(Hydroxymethyl)phenyl)-4-methylbenzenesulfonamideTo a solution of (2-aminophenyl)methanol (616 mg, 5.00 mmol) and pyridine (0.485 mL, 6.00 mmol) in CH2Cl2 (25 mL) was added a solution of p-toluenesulfonyl chloride (906 mg, 4.75 mmol) in CH2Cl2 (10 mL) at 0°C. After stirring for 6 h, the reaction was quenched with saturated aqueous NH4Cl and the mixture was extracted with CH2Cl2 three times. The combined organic extracts were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure to give sulfonamide as a white solid; Its 1H-NMR spectra data was completely identical with that reported31); 1H-NMR (400 MHz, CDCl3) δ: 7.86 (br s, 1H), 7.65 (d, J=8.4 Hz, 2H), 7.43 (d, J=7.6 Hz, 1H), 7.30–7.19 (m, 3H), 7.13–7.05 (m, 2H), 4.40 (s, 2H), 2.38 (s, 3H), 2.13 (br s, 1H).
N-(2-(Dimethoxymethyl)phenyl)-4-methylbenzenesulfonamide (7c)To the solution of N-(2-(hydroxymethyl)phenyl)-4-methylbenzenesulfonamide (777 mg, 2.80 mmol), N-methylmorphine N-oxide (NMO) (492 mg, 4.20 mmol), 4 Å moleculer sieves (powdered, 1.4 mg), CH2Cl2 (14 mL), and MeCN (1.5 mL) was added tetrapropylammonium perruthenate (TPAP) (49.2 mg, 0.140 mmol). After stirring for 11 h, the reaction mixture was passed through a silica gel pad. The filtrate was concentrated under reduced pressure to give the crude aldehyde. The crude aldehyde was directly subjected to the next reaction without further purification. To a mixture of the above crude aldehyde and trimethyl orthoformate (0.530 mL, 4.80 mmol) in MeOH (10 mL) was added p-TsOH·H2O (60.3 mg, 0.240 mmol). After stirring for 2 h at 70°C, the reaction was quenched with saturated aqueous NaHCO3 and the resulting mixture was extracted with CH2Cl2 three times. The combined organic extracts were washed with brine, dried over Na2SO4, and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (hexanes : AcOEt=8 : 2) to afford acetal 7c (557 mg, 1.73 mmol, 62%) as a white solid; Rf=0.30 (Silica gel, hexanes : AcOEt=8 : 2); mp 63–66°C (hexanes/CH2Cl2); IR (neat) 3310, 2941, 1495, 1338, 1154, 1060, 768, 665 cm−1; 1H-NMR (400 MHz, CDCl3) δ: 8.13 (br s, 1H), 7.67 (d, J=8.4 Hz, 2H), 7.64 (d, J=8.4 Hz, 1H), 7.35–7.18 (m, 4H), 7.06 (dd, J=8.0, 7.7 Hz, 1H), 4.89 (s, 1H), 3.19 (s, 6H), 2.36 (s, 3H); 13C-NMR (100 MHz, CDCl3) δ: 143.6, 136.9, 135.5, 129.7, 129.4, 128.4, 127.2, 127.1, 124.0, 121.4, 103.5, 53.3, 21.4; ESI-MS m/z Calcd for C16H19NNaO4S 344.0927 (M++H). Found 344.0927.
General Procedure for Gold-Catalyzed Quinoline Synthesis2-(4-Methoxyphenyl)quinoline (8a)A 10 mL test tube equipped with a magnetic stirring bar was charged with AgOTf (4.1 mg, 0.016 mmol, 10 mol%), PPh3AuCl (7.9 mg, 0.016 mmol, 10 mol%) and toluene (0.74 mL, 0.25 M). To the solution were added alkyne 12a (93 µL, 0.80 mmol, 5.0 equiv), DTBP (14 µL, 0.064 mmol, 40 mol%), i-PrOH (245 µL, 20 equiv) and amide 7c (51.5 mg, 0.160 mmol) at room temperature. The mixture was allowed to warm to 110°C and the suspension was stirred until TLC (hexanes : AcOEt=8 : 2) indicated a complete consumption of 7c. The reaction mixture was filtered through a pad of Celite and concentrated under reduced pressure. The residue was purified by preparative TLC on silica gel (hexanes : AcOEt=8 : 2, then toluene; hexanes=3 : 1) to afford quinoline 8a (24.8 mg, 0.105 mmol, 66%). 1H-NMR spectra data was completely identical with that reported32); 1H-NMR (400 MHz, CDCl3) δ: 8.21–8.11 (m, 4H), 7.84 (d, J=8.8 Hz, 1H), 7.80 (d, J=8.4 Hz, 1H), 7.71 (dd, J=8.8, 8.4 Hz, 1H), 7.50 (dd, J=8.4, 8.4 Hz, 1H), 7.05 (d, J=9.2 Hz, 2H), 3.89 (s, 3H).
Sub-gram Scale Reaction2-(4-Methoxyphenyl)quinoline (8a)A 30 mL two-necked round-bottom flask with a magnetic stirring bar and Liebig condenser was charged with AgOTf (40.0 mg, 0.156 mmol, 10 mol%), PPh3AuCl (77.0 mg, 0.156 mmol, 10 mol%) and toluene (6.5 mL, 0.25 M). To the solution were added alkyne 12a (900 µL, 7.78 mmol, 5.0 equiv), DTBP (140 µL, 0.622 mmol, 40 mol%), i-PrOH (2.40 mL, 20 equiv) and amide 7c (500 mg, 1.56 mmol) at room temperature. The mixture was allowed to warm to 110°C and the suspension was stirred until TLC (hexanes : AcOEt=8 : 2) indicated a complete consumption of 7c. The reaction mixture was filtered through a pad of Celite and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (hexanes : AcOEt=92 : 8) to afford quinoline 8a (225 mg, 0.956 mmol, 62%).
2-Phenylquinoline (8b)According to the general procedure described for 8a, quinoline 8b was obtained from 7c with phenylacetylene 12b in 32.0 µmol scale (3.7 mg, 18 µmol, 56%); a white solid; Its 1H-NMR spectra data was completely identical with that reported32); 1H-NMR (400 MHz, CDCl3) δ: 8.22 (d, J=8.8 Hz, 1H), 8.20–8.13 (m, 3H), 7.88 (d, J=8.4 Hz, 1H), 7.83 (d, J=8.0 Hz, 1H), 7.73 (d, J=8.4, 8.4 Hz, 1H), 7.59–7.42 (m, 4H).
2-(p-Tolyl)quinoline (8c)According to the general procedure described for 8a, quinoline 8c was obtained from 7c with (4-methylphenyl)acetylene 12c in 32.0 µmol scale (4.8 mg, 22 µmol, 68%); a yellow solid; Its 1H-NMR spectra data was completely identical with that reported32); 1H-NMR (400 MHz, CDCl3) δ: 8.20 (d, J=8.8 Hz, 1H), 8.16 (d, J=8.4 Hz, 1H), 8.07 (d, J=8.0 Hz, 2H), 7.87 (d, J=8.4 Hz, 1H), 7.81 (d, J=8.8 Hz, 1H), 7.72 (d, J=8.4, 7.6 Hz, 1H), 7.51 (dd, J=8.4, 7.6 Hz, 1H), 7.33 (d, J=8.0 Hz, 2H), 2.44 (s, 3H).
2-(4-Bromophenyl)quinoline (8d)According to the general procedure described for 8a, quinoline 8d was obtained from 7c with alkyne 12d in 32.0 µmol scale (6.1 mg, 22 µmol, 67%); a yellow solid; Its 1H-NMR spectra data was completely identical with that reported32); 1H-NMR (400 MHz, CDCl3) δ: 8.23 (d, J=8.8 Hz, 1H), 8.15 (d, J=8.8 Hz, 1H), 8.06 (d, J=8.4 Hz, 2H), 7.84 (d, J=8.8 Hz, 1H), 7.83 (d, J=8.8 Hz, 1H), 7.74 (dd, J=8.8, 7.6 Hz, 1H), 7.65 (d, J=8.4 Hz, 2H), 7.54 (dd, J=8.8, 7.6 Hz, 1H).
2-(3,4-Dimethoxyphenyl)quinoline (8e)According to the general procedure described for 8a, quinoline 8e was obtained from 7c with alkyne 12e in 32.0 µmol scale (4.9 mg, 19 µmol, 58%); a white solid; Its 1H-NMR spectra data was completely identical with that reported34); 1H-NMR (400 MHz, CDCl3) δ: 8.18 (d, J=8.8 Hz, 1H), 8.15 (d, J=8.4 Hz, 1H), 7.90 (d, J=2.4 Hz, 1H), 7.85 (d, J=8.8 Hz, 1H), 7.81 (d, J=8.0 Hz, 1H), 7.72 (dd, J=8.4, 8.0 Hz, 1H), 7.67 (dd, J=8.8, 2.4 Hz, 1H), 7.51 (dd, J=8.0, 8.0 Hz, 1H), 7.00 (d, J=8.8 Hz, 1H), 4.06 (s, 3H), 3.97 (s, 3H).
Methyl 4-(Quinolin-2-yl)benzoate (8f)According to the general procedure described for 8a, quinoline 8f was obtained from 7b with alkyne 12f in 32.0 µmol scale (2.6 mg, 6.3 µmol, 31%); a white solid; Its 1H-NMR spectra data was completely identical with that reported33); 1H-NMR (400 MHz, CDCl3) δ: 8.30–8.17 (m, 6H), 7.92 (d, J=8.8 Hz, 1H), 7.85 (d, J=8.4 Hz, 1H), 7.76 (ddd, J=8.4, 8.4, 1.6 Hz, 1H), 7.57 (ddd, J=8.4, 8.4, 1.6 Hz, 1H), 3.97 (s, 3H).
2-(4-Nitrophenyl)quinoline (8g)According to the general procedure described for 8a, quinoline 8g was obtained from 7c with alkyne 12g in 32.0 µmol scale (2.8 mg, 11 µmol, 35%); a yellow solid; Its 1H-NMR spectra data was completely identical with that reported; 1H-NMR (400 MHz, CDCl3) δ: 8.42–8.33 (m, 4H), 8.31 (d, J=8.4 Hz, 1H), 8.19 (d, J=8.4 Hz, 1H), 7.94 (d, J=8.4 Hz, 1H), 7.88 (d, J=8.4 Hz, 1H), 7.79 (dd, J=8.4, 7.6 Hz, 1H), 7.60 (dd, J=8.4, 7.6 Hz, 1H).
2-(Naphthalen-1-yl)quinoline (8h)According to the general procedure described for 8a, quinoline 8h was obtained from 7cw with alkyne 12h in 32.0 µmol scale (3.2 mg, 13 µmol, 39%); an orange oil; Its 1H-NMR spectra data was completely identical with that reported32); 1H-NMR (400 MHz, CDCl3) δ: 8.30 (d, J=8.4 Hz, 1H), 8.23 (d, J=8.8 Hz, 1H), 8.13 (d, J=8.4 Hz, 1H), 7.99–7.89 (m, 3H), 7.82 (dd, J=8.4, 7.2 Hz, 1H), 7.75–7.69 (m, 2H), 7.64–7.56 (m, 2H), 7.55–7.44 (m, 2H).
This work was financially supported by the Cabinet Office, Government of Japan through its “Funding Program for Next Generation World-Leading Researchers (LS008), JSPS KAKENHI Grant Numbers JP26253001 and JP24790003, and the Platform Project for Supporting in Drug Discovery and Life Science Research(Platform for Drug Discovery, Informatics, and Structural Life Science)from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan and Japan Agency for Medical Research and development (AMED).
The authors declare no conflict on interest.