Experimental
General RemarksThe reactions were performed in an oven-dried test tube or round bottom flask with a Teflon-coated magnetic stirring bar unless otherwise noted. All work-up and purification procedures were carried out with reagent-grade solvents under ambient atmosphere. Infrared (IR) spectra were recorded on a JASCO FT/IR 4100 Fourier transform infrared spectrophotometer. NMR was recorded on JEOL ECS-400 (1H-NMR: 400 MHz, 13C-NMR: 100 MHz) or on Bruker AVANCE 500 (1H-NMR: 500 MHz, 13C-NMR: 125 MHz) spectrometers. Chemical shifts for proton are reported in parts per million downfield from tetramethylsilane and are referenced to residual protium in the NMR solvent (CDCl3: δ 7.26 ppm, CD3OD: δ 3.30 ppm, DMSO-d6: 2.49 ppm). For 13C-NMR, chemical shifts were reported in the scale relative to NMR solvent (CDCl3: δ 77.0 ppm, CD3OD: δ 49.0 ppm, DMSO-d6: 39.7 ppm) as an internal reference, or calibrated based on independently recorded peak of 3-trimethylsilylpropanoic acid (TSP) at 0 ppm in D2O. NMR data are reported as follows: chemical shifts, multiplicity (s: singlet, d: doublet, dd: doublet of doublets, t: triplet, m: multiplet, br: broad signal), coupling constant (Hz), and integration. Optical rotation was measured using a 2 mL cell with a 1.0 dm path length on a JASCO polarimeter P-1030. High-resolution (HR)-mass spectra (ESI-Orbitrap) were measured on Thermo Fisher Scientific LTQ Orbitrap XL. Unless otherwise noted, materials were purchased from commercial suppliers and were used without purification. For reaction, tetrahydrofuran (THF), N,N-dimethylformamide (DMF), CH3CN, and CH2Cl2 were purified by passing through a solvent purification system (Glass Contour). Dry MeOH was purchased from Wako Pure Chemical Industries, Ltd. (Osaka, Japan) and used as received.
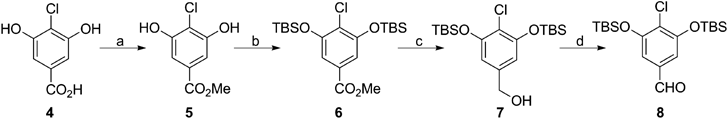
Methyl 4-Chloro-3,5-dihydroxybenzoate (5)To a solution of 4 (3.00 g, 15.9 mmol) in 15 mL of MeOH was added dropwise 0.3 mL of H2SO4 at 0°C. The mixture was stirred at room temperature (r.t.) for 3.5 h, and half of MeOH was removed under reduced pressure and followed by addition of saturated NaHCO3 until the pH value of the mixture reached to around 7. The resultant white solid was collected on funnel and washed thoroughly with H2O, then n-hexane to give 5 as a white powder (2.86 g, 14.1 mmol) in 89% yield; mp 145–146°C; IR (MeOH) ν 3419, 3338, 2360, 1705, 1429, 1376, 1041, 764 cm−1; HR-MS electrospray ionization (ESI) Anal. Calcd for C8H6ClO4 m/z 200.9955 [M−H]−; Found 200.9964; 1H-NMR (500 MHz, CD3OD) δ: 7.06 (2H, s), 3.85 (3H, s); 13C-NMR (125 MHz, CD3OD) δ: 168.1, 155.7, 130.2, 114.6, 109.1, 52.7
Methyl 3,5-Bis((tert-butyldimethylsilyl)oxy)-4-chlorobenzoate (6)To a solution of 5 (2.86 g, 14.1 mmol) in 70 mL of CH2Cl2 was added imidazole (4.23 g, 62.1 mmol), and TBSCl (4.68 g, 31.1 mmol), successively at 0°C. The mixture was stirred at r.t. for 4 h. The reaction mixture was diluted with CHCl3, and washed with saturated NH4Cl, and brine successively. The combined organic layers were dried over Na2SO4, and was concentrated to dryness. The residue was purified with silica gel column chromatography (n-hexane/AcOEt=6/1) to give 6 as a white powder (5.80 g, 13.5 mmol) in 95% yield; mp 95–96°C; IR (CHCl3) ν 2930, 2859, 1724, 1434, 1361, 1253, 841, 783 cm−1; HR-MS (ESI) Anal. Calcd for C20H36ClO4Si2 m/z 431.1841 [M+H]+; Found 431.1822; 1H-NMR (500 MHz, CD3OD) δ: 7.21 (2H, s), 3.89 (3H, s), 1.06 (18H, s), 0.25 (12H, s); 13C-NMR (125 MHz, CD3OD) δ: 167.5, 154.3, 130.0, 124.3, 115.3, 53.0, 26.2, 19.3, −4.3.
(3,5-Bis((tert-butyldimethylsilyl)oxy)-4-chlorophenyl)methanol (7)To a solution of 6 (5.80 g, 13.5 mmol) in 90 mL of CH2Cl2 was added DIBAL-H (diisobutylalminium hydride, 1.0 M in n-hexane, 26.9 mL, 26.9 mmol) at −78°C, and the mixture was stirred for 1 h at the same temperature. Then, 90 mL of aqueous solution of potassium sodium tartrate was added, and extracted with CHCl3. The organic layer was dried over Na2SO4 and concentrated in vacuo to afford 7, as a colorless oil (5.40 g, 13.4 mmol) in quantitative yield; IR (KBr) ν 3327, 2931, 2860, 1577, 1432, 1254, 1098, 829 cm−1; HR-MS (ESI) Anal. Calcd for C19H36ClO3Si2 m/z 403.1892 [M+H]+; Found 403.1882; 1H-NMR (500 MHz, CD3OD) δ: 6.60 (2H, s), 4.48 (2H, s), 1.05 (18H, s), 0.23 (12H, s); 13C-NMR (125 MHz, CD3OD) δ: 154.1, 142.4, 117.2, 113.0, 64.4, 26.2, 19.3, −4.2.
3,5-Bis((tert-butyldimethylsilyl)oxy)-4-chlorobenzaldehyde (8)To a solution of 7 (1.00 g, 2.48 mmol) in 12.4 mL of CH2Cl2 was added PCC (pyridinium chlorochromate, 588 mg, 2.73 mmol), and silica gel (588 mg) at 0°C, and the mixture was stirred for 1 h at r.t. Then, PCC (294 mg, 1.36 mmol) was added and stirred for additional 1 h. The reaction mixture was poured onto the pad of Celite, washed with CHCl3 thoroughly, and the filtrate was concentrated in vacuo. The organic layer was died over Na2SO4, and was concentrated to dryness. The residue was purified with silica gel column chromatography (n-hexane/AcOEt=8/1) to give 8 as a colorless oil (830 mg, 2.07 mmol) in 83% yield; IR (KBr) ν 2931, 2860, 1703, 1574, 1433, 1350, 827 cm−1; HR-MS (ESI) Anal. Calcd for C19H34ClO3Si2 m/z 401.1735 [M+H]+; Found 401.1728; 1H-NMR (400 MHz, CDCl3) δ: 9.83 (1H, s), 7.01 (2H, s), 1.05 (18H, s), 0.26 (12H, s); 13C-NMR (100 MHz, CDCl3) δ: 191.1, 153.8, 135.0, 125.5, 114.0, 113.3, 25.8, 18.5, −4.2.
tert-Butyl 2-((S)-2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-3-hydroxy-3-methylbutanamido)-2-(dimethoxyphosphoryl)acetate (11)To a mixture of 9 (1.09 g, 3.06 mmol), and 10 (1.10 g, 4.60 mmol) in 10 mL of THF was added DMT-MM (1.27 g, 4.60 mmol), and the solution was stirred at r.t. for 5 h. The reaction was quenched with H2O, AcOEt was added to the mixture for extraction, and the combined organic layers were concentrated to dryness. The residue was suspended in toluene and concentrated, which was repeated 5 times. The residue was purified with silica gel column chromatography (CH2Cl2/AcOEt=1/1) to give 11 as a colorless oil (1.01 g, 1.75 mmol) in 57% yield as a colorless oil; IR (KBr) ν 3301, 2979, 1731, 1666, 1525, 1248, 1154, 1041, 758 cm−1; HR-MS (ESI) Anal. Calcd for C28H37N2NaO9P m/z 599.2134 [M+Na]+; Found 599.2123; 1H-NMR (500 MHz, CD3OD) δ: 7.79 (2H, d, J=7.6 Hz), 7.67 (2H, m), 7.38 (2H, m), 7.30 (2H, m), 4.43–4.36 (2H, m), 4.24–4.22 (2H, m), 3.85–3.74 (6H, m), 1.49 (4.5 H, s), 1.48 (4.5 H, s), 1.27 (1.5H, s), 1.27 (1.5H, s), 1.22 (1.5H, s), 1.22 (1.5H, s); 13C-NMR (125 MHz, CD3OD) δ: 172.6 (d, J=6.3 Hz), 172.4 (d, J=6.3 Hz), 166.3, 166.2, 158.7, 145.4, 145.2, 142.3, 128.8, 128.2, 126.3, 121.0, 84.9, 79.5, 72.8, 68.1, 63.72, 63.67, 55.0–54.7 (m, overlap), 48.6 (overlapped with solvent peaks), 28.2, 28.1, 27.7, 27.6, 26.1.
tert-Butyl (10S,14S)-10-((tert-Butoxycarbonyl)amino)-17-(dimethoxyphosphoryl)-14-(2-hydroxypropan-2-yl)-2,2-dimethyl-4,12,15-trioxo-3-oxa-5,13,16-triazaoctadecan-18-oate (13)To a solution of 11 (91.0 mg, 0.158 mmol) in 39 mL of DMF was added 17 µL of piperidine (0.174 mmol), and the mixture was stirred at r.t. for 30 min. Then, the solution was concentrated to dryness. The resultant residue was dissolved in 1.5 mL of MeOH, to which were added 12 (62.7 mg, 0.174 mmol), DMT-MM (52.4 mg, 0.189 mmol), and the resultant mixture was stirred at r.t. for 3 h. The solution was diluted with AcOEt, washed with 1 M hydrochloric acid, and the organic layer was dried over Na2SO4 and concentrated in vacuo. The residue was purified with silica gel column chromatography (CHCl3/MeOH=20/1) to give 13 (44.0 mg, 40%) as a colorless oil (mixture of diastereomer); IR (KBr) ν 3324, 2979, 1688, 1522, 1251, 1171, 1038, 755 cm−1; HR-MS (ESI) Anal. Calcd for C30H57N4NaO12P m/z 719.3608 [M+Na]+; Found 719.3584; 1H-NMR (400 MHz, CDCl3) δ: 7.29 (1H, br), 6.74 (0.5H, br), 6.69 (0.5H, d, J=8.6 Hz), 5.12–4.99 (1H, m), 4.70 (1H, br), 4.39 (1H, dd, J=8.3, 4.2 Hz), 3.84–3.80 (6H, m), 3.80–3.69 (2H, m), 3.10 (1H, br), 2.49 (2H, m), 1.85–1.37 (6H, m), 1.50 (4.5H, s), 1.49 (4.5H, s), 1.47 (9H, s), 1.43 (4.5H, s), 1.42 (4.5H, s), 1.34 (1.5H, s), 1.33 (1.5H, s), 1.22 (1.5H, s), 1.21 (1.5H, s); 13C-NMR (125 MHz, CDCl3) δ: 171.7, 170.8 (d, J=6.3 Hz), 170.7 (d, J=5.0 Hz), 165.1, 164.9, 156.2, 155.7, 84.2, 84.0, 79.3, 79.0, 71.7, 59.5, 59.2, 58.5, 54.1 (d, J=6.3 Hz), 54.0 (d, J=6.3 Hz), 51.0 (d, J=144 Hz), 50.8 (d, J=145 Hz), 48.5, 40.8, 40.2, 34.0, 29.5, 28.4, 27.8, 27.3, 25.7, 25.4, 23.3, 18.4.
tert-Butyl (10S,14S)-10-((tert-Butoxycarbonyl)amino)-17-((Z)-4-chloro-3,5-dihydroxybenzylidene)-14-(2-hydroxypropan-2-yl)-2,2-dimethyl-4,12,15-trioxo-3-oxa-5,13,16-triazaoctadecan-18-oate (14)To a solution of 13 (150 mg, 0.215 mmol) in 1 mL of CH2Cl2 was added DBU (36.0 µL, 0.235 mmol), and 8 (78.0 mg, 0.195 mmol) at r.t. successively. The mixture was stirred at the same temperature for 3 h, diluted with CH2Cl2, washed with brine, dried over Na2SO4, and concentrated. The resultant residue was partially purified with silica gel column chromatography (CHCl3/MeOH=20/1) to give coupling product as a mixture of cis- and trans-isomers, and partially desilylated compounds. Then, this material was dissolved in 4 mL of CH2Cl2, to which was added 1.0 M TBAF in THF (0.47 mL, 0.470 mmol) at r.t. The solution was stirred for 40 min at the same temperature, diluted with CHCl3, washed with saturated NH4Cl and brine, successively. The organic layer was dried over Na2SO4, and concentrated to dryness. The resultant residue was partially purified with silica gel column chromatography (CHCl3/MeOH=20/1) to give 14 as a colorless oil (50.4 mg, 67.9 µmol) in 26% yield over 2 steps with concomitant formation of E-isomer (13.2 mg, 17.8 µmol, 7%) as a colorless oil; [α]D24 +78.6 (c 0.71, MeOH); IR (KBr) ν 3306, 1977, 1686, 1510, 1367, 1280, 1254, 1165 cm−1; HR-MS (ESI) Anal. Calcd for C35H55N4NaO11P m/z 765.3454 [M+Na]+; Found 765.3442; 1H-NMR (500 MHz, CDCl3) δ: 7.97 (2H, br), 7.18 (1H, s), 6.80 (2H, br), 4.90 (1H, br), 4.69 (1H, m), 4.28 (1H, m), 3.73 (1H, br), 3.03 (2H, m), 2.60–2.40 (2H, m), 1.52 (9H, s), 1.45–1.16 (6H, m), 1.44 (9H, s), 1.37 (9H, s), 1.37 (3H, s), 1.30 (3H, s); 13C-NMR (125 MHz, CDCl3) δ: 173.5, 170.5, 163.9, 156.4, 156.1, 153.0, 132.3, 132.1, 126.0, 109.6, 109.4, 82.5, 80.1, 79.4, 71.2, 61.8, 48.8, 42.4, 40.1, 34.7, 29.7, 28.5, 28.3, 28.1, 27.4, 26.4, 23.0.
Resormycin (3)To a solution of 14 (22.3 mg, 30.0 µmol) in 0.5 mL of CH2Cl2 was added TFA (trifluoroacetic acid, 0.3 mL) at 0°C. The mixture was stirred at r.t. for 30 min. Then the mixture was concentrated to dryness to give 3 as a white powder (11.3 mg, 15.7 µmol, 52%); [α]D24 +127.7 (c 0.44, MeOH), lit. for HCl salt, [α]D23 +146.8 (c 1, MeOH)8); IR (KBr) ν 3026, 1648, 1584, 1428, 1391, 1205, 1004 cm−1; HR-MS (ESI) Anal. Calcd for C21H32N4O7 m/z 487.1960 [M+H]+; Found 487.1941; 1H-NMR (500 MHz, DMSO-d6) δ: 8.51 (1H, s), 8.50 (1H, s), 8.03 (3H, s), 7.87 (3H, s), 7.00 (1H, s), 6.77 (2H, m), 5.06 (1H, br), 4.53 (1H, d, J=9.0 Hz), 3.39 (1H, m), 2.75–2.57 (3H, m), 1.62–1.36 (6H, m), 1.23 (3H, s), 1.22 (3H, s); 13C-NMR (125 MHz, DMSO-d6) δ: 169.9, 169.6, 166.1, 154.0, 132.0, 130.9, 126.7, 108.6, 108.5, 71.4, 60.4, 47.9, 38.4, 37.3, 31.4, 27.1, 26.4, 26.1, 21.3.
(S)-7-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-3-((tert-butoxycarbonyl)amino)heptanoic Acid (16)To a solution of 15 (49.2 mg, 0.123 mmol) in 0.6 mL of MeOH was added 10% palladium carbon (Pd/C), and the resulting suspension was stirred under atmospheric pressure of H2 at r.t. for 5 h. After removal of the catalyst by filtration on a pad of Celite, the filtrate was concentrated to dryness to give Boc-L-β-homolysine. This material was dissolved in 1/1 mixture of CH3CN and H2O, to which were added NaHCO3 (21.0 mg, 0.246 mmol), and Fmoc-Cl (9-fluorenylmethyl chloroformate, 35.0 mg, 0.135 mmol), successively. The mixture was stirred at r.t. for 1.5 h. Then, the solution was quenched with 1 M hydrochloric acid (until pH was adjusted to around 3.5), extracted with AcOEt. The combined organic layers were washed with brine, dried over Na2SO4, and concentrated in vacuo. The residue was purified with preparative TLC (CHCl3/MeOH=15/1) to give 16 as a colorless amorphous (40.9 mg, 84.8 µmol) in 69% yield; [α]D24 −2.47 (c 1.8, MeOH); IR (KBr) ν 1694, 1523, 1450, 1250, 1169, 741 cm−1; HR-MS (ESI) Anal. Calcd for C27H34N2NaO6 m/z 505.2315 [M+Na]+; Found 505.2301; 1H-NMR (500 MHz, CD3OD) δ: 7.79 (1H, d, J=7.5 Hz), 7.65 (1H, d, J=7.5 Hz), 7.40 (1H, dd, J=7.5, 7.5 Hz), 7.32 (1H, dd, J=7.5, 7.5 Hz), 4.34 (2H, d, J=6.9 Hz), 4.19 (1H, t, J=6.9 Hz), 3.85 (1H, br), 3.11 (2H, t, J=6.8 Hz), 2.38 (2H, m), 1.59–1.30 (6H, m), 1.42 (9H, s); 13C-NMR (125 MHz, CD3OD) δ: 177.4, 158.9, 157.9, 145.4, 142.6, 128.8, 128.2, 126.2, 121.0, 79.8, 67.6, 42.3, 41.7, 35.6, 30.7, 28.8, 24.4.
tert-Butyl (9S,13S)-9-((tert-Butoxycarbonyl)amino)-16-(dimethoxyphosphoryl)-1-(9H-fluoren-9-yl)-13-(2-hydroxypropan-2-yl)-3,11,14-trioxo-2-oxa-4,12,15-triazaheptadecan-17-oate (18)To a solution of 11 (2.26 g, 3.93 mmol) in 39 mL of DMF was added 428 µL of piperidine (4.32 mmol), and the mixture was stirred at r.t. for 10 min. Then, the solution was concentrated to dryness. The resultant residue (17) was dissolved in 10 mL of THF, to which were added 16 (948 mg, 1.97 mmol), DMT-MM (1.09 g, 3.93 mmol), and the resultant mixture was stirred at r.t. for 5 h. The solution was diluted with AcOEt, washed with 1 M hydrochloric acid, and the organic layer was dried over Na2SO4 and concentrated in vacuo. The residue was purified with silica gel column chromatography (n-hexane/AcOEt=2/1, CHCl3 only, CHCl3/MeOH=20/1) to give 18 (1.22g, 76%) as a white powder (mixture of diastereomer); IR (KBr) ν 3308, 2975, 2934, 2360, 1693, 1522, 1250, 1156, 1035 cm−1; HR-MS (ESI) Anal. Calcd for C40H59N4NaO12P m/z 841.3765 [M+Na]+; Found 841.3750; 1H-NMR (500 MHz, CD3OD) δ: 7.79 (1H, d, J=7.5 Hz), 7.64 (1H, d, J=7.5 Hz), 7.39 (1H, dd, J=7.5, 7.5 Hz), 7.30 (1H, dd, J=7.5, 7.5 Hz), 4.53 (1H, s), 4.34 (2H, d, J=6.6 Hz), 4.19 (1H, t, J=6.6 Hz), 3.86 (1H, br), 3.83–3.79 (6H, m), 3.10 (2H, br), 2.45–2.40 (2H, m), 1.58–1.20 (6H, m), 1.48 (9H, s), 1.41 (9H, br), 1.28 (1.5H, s), 1.28 (1.5H, s), 1.23 (1.5H, s), 1.23 (1.5H, s); 13C-NMR (125 MHz, CD3OD) δ: 173.6, 172.2 (d, J=6.3 Hz), 172.0 (d, J=6.3 Hz), 166.4, 166.2, 145.4, 142.7, 128.8, 128.2, 126.2, 121.0, 84.9, 80.1, 79.5, 72.9, 72.8, 67.6, 61.6, 55.0 (d, J=7.5 Hz), 54.84 (d, J=6.3 Hz), 54.76 (d, J=7.5 Hz), 49.6 (overlapped with solvent peaks), 42.6, 42.5, 41.6, 35.5, 30.7, 28.9, 28.2, 27.8, 27.7, 26.2, 26.1, 24.2.
tert-Butyl N5-((5S)-7-(((2S)-1-((2-(tert-Butoxy)-1-(dimethoxyphosphoryl)-2-oxoethyl)amino)-3-hydroxy-3-methyl-1-oxobutan-2-yl)amino)-5-((tert-butoxycarbonyl)amino)-7-oxoheptyl)-N2-(tert-butoxycarbonyl)-L-glutaminate (19)To a solution of 18 (700 mg, 0.855 mmol) in 7 mL of DMF was added 93 µL of piperidine (0.940 mmol), and the mixture was stirred at r.t. for 30 min. Then, the solution was concentrated to dryness. The resultant residue was dissolved in 2 mL of THF, to which were added Boc-L-Glu-OtBu (130 mg, 0.427 mmol), DMT-MM (237 mg, 0.855 mmol), and the resultant mixture was stirred at r.t. for 18 h. The solution was diluted with AcOEt, washed with 1 M hydrochloric acid, and the organic layer was dried over Na2SO4 and concentrated in vacuo. The residue was purified with silica gel column chromatography (CHCl3/MeOH=10/1) to give 19 (307 mg, 81%) as a colorless oil (mixture of diastereomer); IR (KBr) ν 3309, 2978, 1691, 1654, 1525, 1367, 1253, 1156, 1037 cm−1; HR-MS (ESI) Anal. Calcd for C39H72N5NaO15P m/z 904.4660 [M+Na]+; Found 904.4662; 1H-NMR (500 MHz, CD3OD) δ: 4.53 (1H, s), 3.95 (1H, m), 3.88 (1H, m), 3.84–3.81 (6H, m), 3.15 (2H, t, J=6.8 Hz), 2.43 (2H, d, J=6.4 Hz), 2.26 (2H, t, J=6.4 Hz), 2.06 (1H, m), 1.85 (1H, m), 1.54–1.34 (4H, m), 1.49 (9H, s), 1.46 (9H, s), 1.44 (9H, s), 1.42 (9H, br), 1.28 (3H, s), 1.23 (1.5H, s), 1.23 (1.5H, s); 13C-NMR (125 MHz, CD3OD) δ: 174.8, 173.5, 173.3, 172.2 (d, J=6.3 Hz), 171.9 (d, J=6.3 Hz), 166.4, 166.2, 158.1, 157.9, 84.9, 61.6, 61.5, 55.0 (d, J=7.5 Hz), 55.0–54.7 (m, overlap), 49.7 (overlapped with solvent peaks), 42.5, 42.4, 40.4, 35.5, 33.4, 30.2, 28.9, 28.8, 28.3, 28.2, 27.8, 27.7, 26.2, 26.1, 24.4.
tert-Butyl N5-((S)-7-(((S)-1-(((Z)-3-(tert-Butoxy)-1-(4-chloro-3,5-dihydroxyphenyl)-3-oxoprop-1-en-2-yl)amino)-3-methyl-1-oxobutan-2-yl)amino)-5-((tert-butoxycarbonyl)amino)-7-oxoheptyl)-N2-(tert-butoxycarbonyl)-L-glutaminate (20)To a solution of 19 (15.8 mg, 17.9 µmol) in 0.2 mL of CH2Cl2 was added DBU (4.0 µL, 21.5 µmol), and 8 (7.0 mg, 16.3 µmol) at r.t. successively. The mixture was stirred at the same temperature for 1.5 h, diluted with CH2Cl2, washed with brine, dried over Na2SO4, and concentrated. The resultant residue was partially purified with silica gel column chromatography (CHCl3/MeOH=15/1) to give coupling product as a mixture of cis- and trans-isomers, and partially desilylated compounds. Then, this material was dissolved in 0.25 mL of CH2Cl2, to which was added 1.0 M TBAF in THF (28.0 µL, 28.0 μmol) at r.t. The solution was stirred for 40 min at the same temperature, diluted with CHCl3, washed with saturated NH4Cl and brine, successively. The organic layer was dried over Na2SO4, and concentrated to dryness. The resultant residue was partially purified with silica gel column chromatography (CHCl3/MeOH=15/1) to give 20 as a colorless oil (6.4 mg, 6.89 µmol) in 42% yield over 2 steps with concomitant formation of E-isomer (3.2 mg, 3.45 µmol, 21%) as a colorless oil; [α]D25 –48.4 (c 0.24, MeOH); IR (KBr) ν 3310, 2977, 1697, 1653, 1507, 1367, 1279, 1254, 1158, 1053 cm−1; HR-MS (ESI) Anal. Calcd for C44H71ClN5O14 m/z 928.4686 [M+H]+; Found 928.4664; 1H-NMR (400 MHz, CD3OD) δ: 7.11 (1H, s), 6.74 (2H, s), 4.49 (1H, s), 3.97–3.81 (2H, m), 3.11 (2H, m), 2.48 (2H, d, J=7.6 Hz), 2.25 (2H, d, J=9.2 Hz), 2.20–2.01 (1H, m), 1.90–1.80 (1H, m), 1.52 (9H, s), 1.50–1.25 (6H, m), 1.46 (9H, s), 1.44 (9H, s), 1.40 (9H, s), 1.38 (3H, s), 1.33 (3H, s); 13C-NMR (125 MHz, CD3OD) δ: 174.8, 174.3, 173.3, 172.4, 165.6, 158.1, 158.0, 155.5, 133.6, 133.5, 127.9, 110.8, 110.1, 83.2, 82.8, 80.6, 80.2, 72.4, 62.6, 55.6, 42.8, 40.3, 35.4, 33.4, 30.0, 28.9, 28.8, 28.4, 28.3, 27.5, 27.0, 24.3.
Androprostamine A (1)To a solution of 20 (11.6 mg, 12.5 µmol) in 0.25 mL of CH2Cl2 was added TFA (125 µL) at 0°C. The mixture was stirred at r.t. for 30 min. Then the mixture was concentrated to dryness to give a crude material containing 1, which was purified with LH-20 (MeOH) and quenched with saturated NaHCO3. The 1H-NMR of the resultant sample was identical to that of natural one. The sample was further purified with HPLC (Capcell Pak C18 UG, Shiseido, Tokyo, Japan, 20×250 mm; eluent, 10% CH3CN in H2O containing 0.1% of TFA; detection, UV at 300 nm; flow rate, 10 mL/min) to give androprostamine A TFA salt as a colorless amorphous. The material was dissolved in H2O, to which 1 M NH4OH was added until the pH reached to around 9. The solution was concentrated and the resulting crude material was purified by LH-20 (MeOH) to give 1 (1.9 mg, 2.05 µmol, 25%); [α]D27 +42.4 (c 0.085, MeOH), (natural sample: [α]D27 +43.2 (c 0.085, MeOH)); IR (KBr) ν 3100, 1671, 1397, 1202, 1136 cm−1; HR-MS (ESI) Anal. Calcd for C26H39ClN5O10 m/z 616.2385 [M+H]+; Found 616.2377; 1H-NMR (500 MHz, D2O) δ: 7.20 (1H, s), 6.75 (2H, s), 4.49 (1H, s), 3.75 (1H, t, J=6.5 Hz), 3.60 (1H, m), 3.06 (2H, m), 2.78 (1H, dd, J=14.4, 5.0 Hz), 2.74 (1H, dd, J=14.4, 6.0 Hz), 2.39 (1H, m), 2.12 (1H, m), 1.60 (1H, m), 1.53 (1H, m), 1.35 (3H, s), 1.36–1.32 (4H, m), 1.33 (3H, s); 13C-NMR (125 MHz, D2O) δ: 177.2, 176.7, 174.9, 174.1, 173.2, 155.4, 136.7, 133.4, 132.5, 111.7, 111.3, 74.5, 64.3, 56.9, 51.6, 41.7, 38.7, 34.3, 33.9, 30.6, 29.3, 28.5, 27.9, 24.7.