Regular Articles
Convergent Synthesis of Dronedarone, an Antiarrhythmic Agent
2016 年 64 巻 8 号 p. 1149-1153
詳細
2016 年 64 巻 8 号 p. 1149-1153
We have developed a convergent synthesis of dronedarone, an antiarrhythmic agent. The key steps of the process are the construction of a benzofuran skeleton by iodocyclization and the carbonylative Suzuki–Miyaura cross-coupling for biaryl ketone formation. This synthetic route required only eight steps from 2-amino-4-nitrophenol in 23% overall yield.
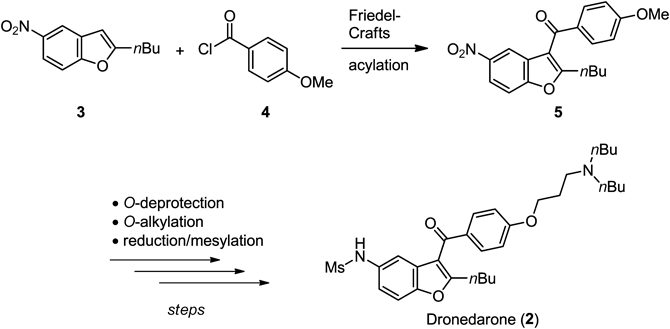
Atrial fibrillation is one of the most common types of arrhythmia, and is a disease of irregular and rapid heart rhythm. As atrial fibrillation may induce the formation of a thrombus at the atrium, it increases the risk of cardiogenic cerebral embolism. To maintain normal sinus rhythm for the patients with atrial fibrillation, a controlled electric shock and drug therapy are used. Amiodarone (1; Fig. 1) is widely used as a class III antiarrhythmic agent and is an effective multichannel blocker. However, amiodarone has iodo-substituents and is lipophilic, it causes thyroid dysfunction and has a long lifetime up to 100 d that is accompanied by its accumulation in adipose tissue and other organs as non-cardiovascular adverse effects.1–3) Dronedarone (2), whose hydrochloride (marketed as Multaq® from Sanofi-Aventis) was approved by the United States Food and Drug Administration (FDA) in 2009,4,5) is a finely modified analog of amiodarone; loss of iodine functionality prevented thyroid side effects and introduction of a methanesulfonamide on benzo[b]furan reduced lipophilicity, resulting in a half-life of 24 h and lower tissue accumulation.6)

The first synthesis of dronedarone (2) was reported by Gubin et al.,7) and involved a linear process from 2-butyl-5-nitrobenzo[b]furan (3) as a starting material (Chart 1). Friedel–Crafts acylation of 3 with 4-methoxybenzoyl chloride (4) was the key step for biaryl ketone formation. Deprotection from anisole 5 to phenol followed by O-alkylation formed the right segment of 2, and the reduction of the nitro group and mesylation of the free amine provided dronedarone (2). Several other synthetic routes of 2 also depended on the use of benzo[b]furan 3, and minor improvements of reaction conditions and the exchange of steps have been reported.8–13) From a drug-discovery point of view, those processes lack the flexibility of the 2-position in benzo[b]furan because this position was defined at the first stage of the synthesis. Therefore, an alternative route toward 2 that contains the introduction of a variety of substituents at the 2-position on benzo[b]furan at later stage was needed.
We have already developed a versatile construction of 3-iodobenzo[b]furans by electrophilic iodocyclization of ethoxyethyl ethers to alkynes14–16) (Chart 2). Advantages of our synthetic method for benzo[b]furan are as follows: (1) ethoxyethyl ether serves as not only protecting group for the preparation of precursors, but also as a good leaving group for iodocyclization; (2) a wide variety of substituents such as aryl, alkyl, and amide groups at the 2-position on benzo[b]furans is allowed; (3) the iodine moiety after cyclization can be easily transformed to other substituents by transition metal-catalyzed cross-coupling reactions; and (4) this iodocyclization is completed within 10 min at room temperature. Therefore, we considered that our methodology could be adapted for the synthesis of dronedarone (2) having a 2-alkylbenzo[b]furan core. Our synthetic plan is shown in Chart 3; biaryl ketone of 2 would be formed at the last stage by carbonylative Suzuki–Miyaura cross-coupling17) with 3-iodobenzo[b]furan 6 and arylboronic acid 7 aiming for a convergent process, and 6 would be derived from alkyne 8 by our developed iodocyclization protocol. Aryl alkyne 8 would be formed by Sonogashira coupling of corresponding iodoarene 9 and 1-hexyne. The iodo moiety of 9 was expected to be accessible from inexpensive 4-nitro-2-aminophenol (10) by a formation of the aryldiazonium salt followed by iodination.

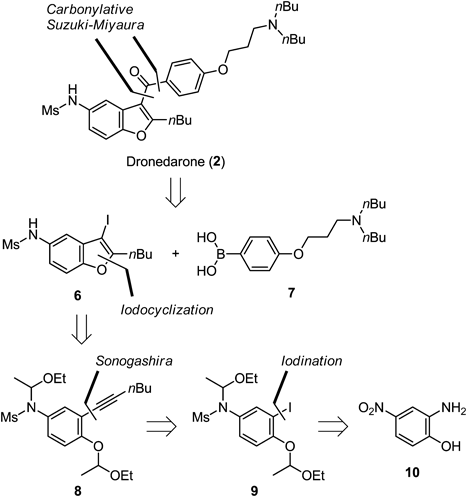
Our synthetic procedure is shown in Chart 4. Aryldiazonium salt formation of 10 and subsequent iodination using potassium iodide afforded iodoarene 11.18,19) The conversion of nitrophenol 11 to 9 was accomplished by a three-step sequence of reduction, N-mesylation, and bis-acetal formation in good yield. Sonogashira cross-coupling of 9 with the terminal alkyne provided 8, a precursor of electrophilic cyclization. According to our iodocyclization protocol,14) exposure of alkyne 8 to the bis(2,4,6-collidine)iodonium hexafluorophosphate [I(coll)2PF6]-BF3·OEt2 combination afforded benzo[b]furan 6 along with its N-ethoxyethyl product. The mixture was then directly treated with hydrochloric acid for hydrolysis to converge 6, a left building block of 2, as a sole product in 77% yield over two steps.

Reagents and conditions: (a) NaNO2, 30% H2SO4, DMSO, 0°C, 1 h; then KI, r.t., 12 h, 77%; (b) (i) SnCl2, EtOH, 70°C, 15 h; (ii) MsCl, pyridine, CH2Cl2, r.t., 15 h; (iii) ethyl vinyl ether, PPTS, CH2Cl2, r.t., 24 h, 72% in 3 steps; (c) 1-hexyne, PdCl2, Ph3P, CuI, Et3N, CH3CN, r.t., 23 h, 95%; (d) (i) I(coll)2PF6, BF3·OEt2, CH2Cl2, r.t., 10 min; (ii) 10% HCl, THF, MeOH, 60°C, 30 min, 77% in 2 steps; (e) nBu2NH, LiBF4, CH3CN, r.t., 4 h, 54%; (f) SOCl2, CHCl3, reflux, 1 h, 89%; (g) 4-bromophenol, Cs2CO3, NaI, CH3CN, 65°C, 23 h, 92%; (h) nBuLi, THF, −78°C, 45 min; then B(OiPr)3, r.t., 23 h; then sat. NH4Cl aq., r.t., 30 min, 46%; (i) PdCl2(PPh3)2, K2CO3, CO (1 atm), anisole, 80°C, 23 h, 57%.
Arylboronic acid 7, a right segment of 2, was prepared by a four-step sequence: nucleophilic ring-opening reaction of oxetane 12 with dibutylamine formed aminoalcohol 13.20) The transformation of the resulting hydroxyl group to the chloro-substituent by thionyl chloride provided 14, and nucleophilic substitution with p-bromophenol constructed alkyl aryl ether 15. Finally, a dihydroxyboryl group was introduced by halogen-lithium exchange reaction of 15 followed by boronic ester formation and subsequent hydrolysis to afford 7.
At the last stage, we studied the carbonylative Suzuki–Miyaura cross-coupling reaction of 6 and 7 for biaryl ketone formation aiming toward dronedarone (2). After several examinations, we have found that employing the classical reaction conditions produced 2 in 57% yield without forming a biaryl, a direct coupling product with 6 and 7.17) The 1H- and 13C-NMR spectra of synthetic 2 agreed with those of the reported data.21) Now, the synthesis of 2 was completed in a convergent manner, and our synthetic route of 2 required only linear eight steps from commercially available material 10 in 23% overall yield.
In summary, we have developed a convergent synthetic process of 2, that was characterized by iodocyclization and carbonylative Suzuki–Miyaura cross-coupling reaction as key steps. In our process, setting the substituent at the 2-position on benzo[b]furan occurred at a later stage than in the previously reported route of 2. Thus, a variety of analogous compounds of 2 whose substituent differed at this positon would be easily prepared from 9 in four steps. In addition, this convergent synthesis was achieved in eight steps from 10 in 23% overall yield. Therefore, we envisage that this synthetic route for 2 will be useful for the research and development of new antiarrhythmic agents.
Melting points were measured by a Yanagimoto micro melting point apparatus. IR spectra were measured on a PerkinElmer Spectrum 100 FT-IR spectrometer using CHCl3. 1H-NMR and 13C-NMR spectra were determined on a Varian Gemini-300 or a Varian Mercury-300 or a Varian VXR-500 superconducting FT-NMR spectrometer, respectively. Chemical shifts (δ) are reported in ppm relative to tetramethylsilane as internal reference (CDCl3: δ=0 ppm for 1H) and residual solvent signal (CDCl3: δ=77 ppm for 13C). J-Values are given in Hz. High-resolution (HR)-MS-(electrospray ionization (ESI)) were acquired on a Thermo Fisher Scientific Exactive Orbitrap mass spectrometer. Column chromatography was performed using Kanto Silica Gel 60 N (spherical, neutral). All reactions were carried out under an argon atmosphere. All reagents were directly used as obtained commercially.
2-Iodo-4-nitrophenol (11)According to the modified procedures by Dai and Lai18) and Zhu et al.,19) to a solution of 2-amino-4-nitrophenol (10) (14.8 g, 96.0 mmol) in 30% H2SO4 (500 mL) and dimethyl sulfoxide (DMSO) (500 mL) was added a solution of NaNO2 (9.94 g, 144 mmol) in water (50 mL) at 0°C. The reaction mixture was stirred at the same temperature for 1 h, after which a solution of KI (47.8 g, 288 mmol) in water (50 mL) was added. After stirring at room temperature (r.t.) for 1 h, another batch of KI (47.8 g, 288 mmol) in water (50 mL) was then added. The reaction mixture was stirred at r.t. for 12 h. After reaction was completed, the mixture was extracted with EtOAc, washed with brine and saturated aqueous solution of NaHSO3, dried over Na2SO4, filtered, and evaporated in vacuo. The residue was purified by flash column chromatography on silica gel eluting with hexane/EtOAc=1 : 1 to give 11 (19.7 g, 77%) as yellow crystals. mp 84–86°C (hexane/EtOAc); IR νmax: 3479, 3020, 1602, 1524, 1341 cm−1; 1H-NMR (300 MHz, CDCl3) δ: 8.60 (d, J=2.4 Hz, 1H), 8.18 (dd, J=9.0, 2.7 Hz, 1H), 7.07 (d, J=9.0 Hz, 1H), 6.23 (br s, 1H); 13C-NMR (75 MHz, CDCl3) δ: 160.4, 141.9, 134.4, 126.1, 114.6, 84.5; HR-ESI-MS Calcd for C6H3INO3 [M−H]− 263.9163. Found 263.9164.
N-(4-(1-Ethoxyethoxy)-3-iodophenyl)-N-(1-ethoxyethyl)methanesulfonamide (9)A mixture of 2-iodo-4-nitrophenol (11) (19.8 g, 74.6 mmol) and SnCl2 (79.0 g, 350 mmol) in EtOH (140 mL) was stirred under argon at 70°C for 15 h. After reaction was completed, the mixture was neutralized with 5% aqueous solution of Na2CO3 at 0°C. The mixture was extracted with EtOAc, washed with brine, dried over Na2SO4, filtered, and evaporated in vacuo to afford crude 4-amino-2-iodophenol (16.5 g), which was used for next reaction without further purification.
To a mixture of crude 4-amino-2-iodophenol (16.5 g) in pyridine (6.23 mL, 77.0 mmol) and CH2Cl2 (175 mL) was added MsCl (5.96 mL, 77.0 mmol) at 0°C, and the mixture was stirred at r.t. for 15 h. After reaction was completed, the mixture was quenched with aqueous 6 M NaOH (700 mL), and extracted with CH2Cl2. The resulting aqueous layer was acidified (pH=2) with aqueous 18% HCl at 0°C, and was extracted with EtOAc. The combined organic layer was dried over Na2SO4, filtered, and evaporated in vacuo to obtain crude N-(4-hydroxy-3-iodophenyl)methanesulfonamide (22.0 g), which was used for next reaction without further purification.
To a mixture of crude N-(4-hydroxy-3-iodophenyl)methanesulfonamide (22.0 g) and pyridinium p-toluenesulfonate (PPTS) (1.76 g, 7.00 mmol) in dry CH2Cl2 (350 mL) was added ethyl vinyl ether (20.1 mL, 210 mmol) and stirred at r.t. for 24 h. After reaction was completed, the mixture was quenched with saturated aqueous solution of NaHCO3, extracted with CH2Cl2, dried over Na2SO4, filtered, and evaporated in vacuo. The residue was purified by flash column chromatography on silica gel eluting with hexane/EtOAc=3 : 1 to give 1 : 1 diastereomer mixture of 9 (24.6 g, 72% in 3 steps) as a yellow oil. IR νmax: 2981, 1590, 1341, 1279, 1164 cm−1; 1H-NMR (300 MHz, CDCl3) δ: 7.78 (d, J=2.4 Hz, 1H), 7.30 (dd, J=8.7, 2.4 Hz, 1H), 7.04 (d, J=8.7 Hz, 1H), 5.52 (q, J=6.0 Hz, 1H), 5.47–5.40 (m, 1H), 3.87–3.52 (m, 4H), 2.98 (s, 3H), 1.58 (d, J=5.1 Hz, 3H), 1.30–1.16 (m, 9H); 13C-NMR (75 MHz, CDCl3) δ: 156.14, 156.12, 141.7, 141.6, 132.33, 132.28, 129.4, 115.3, 115.2, 100.6, 100.5, 87.52, 87.49, 84.9, 63.6, 61.3, 61.1, 39.7, 20.0, 19.9, 19.8, 15.1, 15.0; HR-ESI-MS Calcd for C15H24INNaO5S [M+Na]+ 480.0312. Found 480.0312.
N-(4-(1-Ethoxyethoxy)-3-(hex-1-yn-1-yl)phenyl)-N-(1-ethoxyethyl)methanesulfonamide (8)A mixture of 9 (2.29 g, 5.00 mmol), PdCl2 (22.2 mg, 0.125 mmol), PPh3 (65.6 mg, 0.250 mmol), CuI (34.3 mg, 0.180 mmol), Et3N (2.09 mL, 15.0 mmol), and 1-hexyne (1.15 mL, 10.0 mmol) in dry CH3CN (9.3 mL) was stirred at r.t. for 23 h. After reaction was completed, the mixture was quenched with aqueous 5% NH3. The mixture was extracted with EtOAc, washed with brine, dried over MgSO4, filtered, and evaporated in vacuo. The residue was purified by flash column chromatography on silica gel eluting with hexane/EtOAc=4 : 1 to give 1 : 1 diastereomer mixture of 8 (1.95 g, 95%) as a brown oil. IR νmax: 2935, 2233, 1600, 1340, 1265, 1162 cm−1; 1H-NMR (300 MHz, CDCl3) δ: 7.36 (d, J=2.4 Hz, 1H), 7.21 (dd, J=8.7, 2.7 Hz, 1H), 7.03 (d, J=8.7 Hz, 1H), 5.53 (q, J=6.0 Hz, 1H), 5.46–5.40 (m, 1H), 3.90–3.53 (m, 4H), 2.97 (s, 3H), 2.45 (t, J=6.9 Hz, 2H), 1.64–1.44 (m, 7H), 1.29–1.15 (m, 9H), 0.95 (t, J=7.2 Hz, 3H); 13C-NMR (75 MHz, CDCl3) δ: 157.7, 157.6, 135.8, 135.7, 131.83, 131.77, 128.4, 117.1, 117.0, 116.1, 100.7, 100.5, 95.3, 84.8, 76.0, 63.6, 61.4, 61.3, 39.7, 30.6, 21.8, 20.2, 20.1, 19.8, 19.3, 15.11, 15.08, 13.5; HR-ESI-MS Calcd for C21H34NO5S [M+H]+ 412.2152. Found 412.2148.
N-(2-Butyl-3-iodobenzofuran-5-yl)methanesulfonamide (6)To a mixture of 8 (0.823 g, 2.00 mmol) in dry CH2Cl2 (10.0 mL) was added I(coll)2PF6 (2.06 g, 4.00 mmol) and BF3·OEt2 (0.494 mL, 4.00 mmol) at r.t. and was stirred for 10 min. After reaction was completed, the mixture was quenched with saturated aqueous solution of NaHCO3 and saturated aqueous solution of Na2S2O3. The mixture was extracted with CH2Cl2, dried over Na2SO4 filtered, and evaporated in vacuo, which was used for next reaction without further purification.
This residue was solved in tetrahydrofuran (THF) (36 mL), MeOH (36 mL) and aqueous 10% HCl (36 mL), and the mixture was stirred at 60°C for 30 min. After reaction was completed, the mixture was extracted with EtOAc, washed with brine, dried over Na2SO4, filtered, and evaporated in vacuo. The residue was purified by flash column chromatography on silica gel eluting with hexane/EtOAc=3 : 1 to give 6 (0.606 g, 77% in 2 steps) as light green crystals. mp 101–102°C (hexane/EtOAc); IR νmax: 3371, 3255, 2960, 1619, 1583, 1342, 1152 cm−1; 1H-NMR (300 MHz, CDCl3) δ: 7.37 (dd, J=8.4, 0.6 Hz, 1H), 7.25–7.19 (m, 2H), 7.06 (s, 1H), 3.02 (s, 3H), 2.85 (t, J=7.8 Hz, 2H), 1.78–1.67 (m, 2H), 1.40 (sext, J=7.5 Hz, 2H), 0.96 (t, J=7.5 Hz, 3H); 13C-NMR (75 MHz, CDCl3) δ: 160.9, 152.4, 132.14, 132.11, 119.9, 114.8, 111.8, 62.1, 38.9, 29.8, 27.7, 22.1, 13.7; HR-ESI-MS Calcd for C13H17INO3S [M+H]+ 393.9968. Found 393.9975.
3-(Dibutylamino)propan-1-ol (13)According to the literature,20) to a solution of oxetane (12) (4.88 mL, 75.0 mmol) and dibutylamine (25.5 mL, 150 mmol) in dry CH3CN (75 mL) was added LiBF4 (14.1 g, 150 mmol), and was stirred at r.t. for 4 h. After reaction was completed, the mixture was quenched with aqueous 36% NH3 (300 mL). The mixture was extracted with EtOAc, washed with brine, dried over Na2SO4, filtered, and evaporated in vacuo, and purified by distillation under reduced pressure (boiling point (bp) 97°C/4 mmHg) to give 13 (7.63 g, 54%) as a colorless oil. IR νmax: 3207, 2933 cm−1; 1H-NMR (300 MHz, CDCl3) δ: 5.87 (br s, 1H), 3.80 (t, J=5.1 Hz, 2H), 2.64 (t, J=5.4 Hz, 2H), 2.44–2.38 (m, 4H), 1.72–1.64 (m, 2H), 1.51–1.41 (m, 4H), 1.37–1.24 (m, 4H), 0.92 (t, J=7.2 Hz, 6H); 13C-NMR (75 MHz, CDCl3) δ: 64.9, 55.3, 53.9, 29.0, 27.7, 20.6, 14.0; HR-ESI-MS Calcd for C11H26NO [M+H]+ 188.2009. Found 188.2004.
N-Butyl-N-(3-chloropropyl)butan-1-amine (14)A solution of SOCl2 (2.04 mL, 28.0 mmol) in CHCl3 (2 mL) was slowly added to a solution of 13 (3.75 g, 20.0 mmol) in CHCl3 (3.5 mL) at r.t., and the mixture was stirred under reflux for 1 h. After reaction was completed, the mixture was cooled to r.t., and was evaporated in vacuo. The residue was diluted with aqueous 6 M NaOH (26 mL), and the mixture was extracted with EtOAc, dried over Na2SO4, filtered, and evaporated in vacuo, and purified by distillation under reduced pressure (bp 125–130°C/25 mmHg) to give 14 (3.65 g, 89%) as a colorless oil. IR νmax: 2960 cm−1; 1H-NMR (300 MHz, CDCl3) δ: 3.60 (t, J=6.6 Hz, 2H), 2.53 (t, J=6.6 Hz, 2H), 2.38 (t, J=7.5 Hz, 4H), 1.88 (quint, J=6.6 Hz, 2H), 1.46–1.23 (m, 8H), 0.91 (t, J=7.2 Hz, 6H); 13C-NMR (75 MHz, CDCl3) δ: 54.0, 51.0, 43.5, 30.5, 29.4, 20.7, 14.1; HR-ESI-MS Calcd for C11H25ClN [M+H]+ 206.1670. Found 206.1664.
N-(3-(4-Bromophenoxy)propyl)-N-butylbutan-1-amine (15)To a mixture of 14 (2.06 g, 13.0 mmol) and NaI (0.375 g, 2.50 mmol) in CH3CN (100 mL) was added 4-bromophenol (2.20 g, 10.0 mmol) and Cs2CO3 (5.87 g, 18.0 mmol) at r.t., and the mixture was stirred at 60°C for 23 h. After reaction was completed, the mixture was filtered through Celite, and the filtrate was washed with aqueous 3 M NaOH. The organic layer was dried over Na2SO4, filtered, and evaporated in vacuo. The residue was purified by flash column chromatography on silica gel eluting with CH2Cl2/MeOH=10 : 1 to give 15 (3.16 g, 92%) as a colorless amorphous solid. IR νmax: 2959, 1592, 1245, 1072 cm−1; 1H-NMR (300 MHz, CDCl3) δ: 7.35 (d, J=9.0 Hz, 2H), 6.77 (d, J=9.0 Hz, 2H), 3.96 (t, J=6.3 Hz, 2H), 2.56 (t, J=6.9 Hz, 2H), 2.39 (t, J=7.2 Hz, 4H), 1.88 (quint, J=6.9 Hz, 2H), 1.45–1.22 (m, 8H), 0.89 (t, J=7.2 Hz, 6H); 13C-NMR (75 MHz, CDCl3) δ: 158.2, 132.1, 116.3, 112.5, 66.5, 54.0, 50.4, 29.3, 27.1, 20.7, 14.1; HR-ESI-MS Calcd for C17H29BrNO [M+H]+ 341.1427. Found 342.1416.
(4-(3-(Dibutylamino)propoxy)phenyl)boronic Acid (7)To a mixture of 15 (3.08 g, 9.00 mmol) in dry THF (41 mL) at −78°C was added n-BuLi (1.62 M in hexane, 8.33 mL, 13.5 mmol), and stirred at −78°C for 45 min. Then, triisopropylborate (6.24 mL, 27.0 mmol) was added, and stirred at r.t. for 23 h. After reaction was completed, a saturated aqueous solution of NH4Cl was added to the reaction mixture, and was vigorously stirred at r.t. for 30 min. The mixture was extracted with EtOAc, dried over Na2SO4, filtered, and evaporated in vacuo. The residue was purified by flash column chromatography on silica gel eluting with CH2Cl2/MeOH=9 : 1 to give 7 (1.27 g, 46%) as a yellow amorphous solid. IR νmax: 2959, 1603, 1246 cm−1; 1H-NMR (500 MHz, CDCl3) δ: 8.01–7.71 (br m, 2H), 6.82 (d, J=7.0 Hz, 2H), 3.93 (br s, 2H), 2.69 (br t, J=6.5 Hz, 2H), 2.48 (t, J=7.5 Hz, 4H), 1.94 (br quint, J=6.5 Hz, 4H), 1.47–1.40 (m, 4H), 1.25 (sext, J=7.5 Hz, 4H), 0.87 (t, J=7.5 Hz, 6H); 13C-NMR (125 MHz, CDCl3) δ: 160.9, 135.6, 129.4, 113.5, 65.6, 53.4, 50.4, 28.2, 26.1, 20.5, 13.9; HR-ESI-MS Calcd for C17H31BNO3 [M+H]+ 308.2392. Found 308.2381.
N-(2-Butyl-3-(4-(3-(dibutylamino)propoxy)benzoyl)benzofuran-5-yl)methanesulfonamide (Dronedarone, 2)According to the literature,17) a mixture of 6 (0.149 g, 0.380 mmol), 7 (0.128 g, 0.420 mmol), PdCl2(PPh3)2 (8.0 mg, 0.0114 mmol), and K2CO3 (0.158 g, 1.14 mmol) was filled with CO, and dry anisole (2.3 mL) was added. The mixture was vigorously stirred at 80°C for 23 h. After reaction was completed, the mixture was filtered through Celite. The filtrate was extracted with EtOAc, washed with water followed by brine, dried over Na2SO4, filtered, and evaporated in vacuo. The residue was purified by flash column chromatography on silica gel eluting with CH2Cl2/MeOH=15 : 1 to give 2 (0.120 g, 57%) as a colorless oil. IR νmax: 3369, 3257, 2960, 1725, 1639, 1600, 1339, 1255, 1156 cm−1; 1H-NMR (300 MHz, CDCl3) δ: 7.81 (d, J=9.0 Hz, 2H), 7.44 (dd, J=8.4, 0.9 Hz, 1H), 7.30 (s, 1H), 7.29 (d, J=8.4 Hz, 1H), 6.95 (d, J=9.0 Hz, 2H), 4.10 (t, J=6.3 Hz, 2H), 2.92 (s, 3H), 2.85 (t, J=7.5 Hz, 2H), 2.62 (t, J=6.9 Hz, 2H), 2.44 (t, J=7.2 Hz, 4H), 1.96 (quint, J=6.9 Hz, 2H), 1.73 (quint, J=7.5 Hz, 2H), 1.48–1.23 (m, 10H), 0.89 (t, J=6.0 Hz, 6H), 0.87 (t, J=6.0 Hz, 3H), NH was not observed; 13C-NMR (75 MHz, CDCl3) δ: 190.3, 165.8, 163.2, 151.8, 132.4, 131.7, 131.2, 128.2, 120.2, 116.7, 115.5, 114.3, 111.8, 66.5, 53.9, 50.3, 39.0, 30.0, 29.1, 28.0, 26.9, 22.3, 20.7, 14.1, 13.6; HR-ESI-MS Calcd for C31H45N2O5S [M+H]+ 557.3044. Found 557.3026.
This work was supported in part by JSPS KAKENHI Grant Number 15K07878 (T.O.).
The authors declare no conflict of interest.
The online version of this article contains supplementary materials.