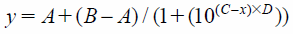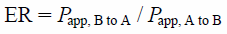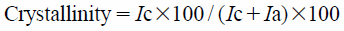Experimental
General Chemistry InformationAll solvents and reagents were obtained from commercial sources and were used as received. Microwave-assisted reactions were carried out in a single-mode reactor, Biotage Initiator 2.0 or 2.5 microwave synthesizer. Yields were not optimized. All reactions were monitored by TLC analysis on Merck Kieselgel 60 F254 plates or Fuji Silysia NH plates, or LC-MS analysis. LC-MS analysis was performed on a Shimadzu LC-MS system operating in atmospheric pressure chemical ionization (APCI) (+ or −) or electrospray ionization (ESI) (+ or −) ionization mode. Analytes were eluted using a linear gradient with a mobile phase of water/acetonitrile containing 0.05% trifluoroacetic acid (TFA) or 5 mM ammonium acetate and detected at 220 nm. Column chromatography was carried out on silica gel ((Merck Kieselgel 60, 70–230 mesh, Merck) or (Chromatorex NH-DM 1020, 100–200 mesh, Fuji Silysia Chemical, Ltd.)), or on prepacked Purif-Pack columns (SI or NH, particle size: 60 µm, Fuji Silysia Chemical, Ltd.). Analytical HPLC was performed using a Corona Charged Aerosol Detector or photo diode array detector with a Capcell Pak C18AQ (3.0 mm ID×50 mm L, Shiseido, Japan) or L-column2 ODS (2.0 mm ID×30 mm L, CERI, Japan) column at a temperature of 50°C and a flow rate of 0.5 mL/min. Mobile phases A and B under neutral conditions were a mixture of 50 mmol/L ammonium acetate, water, and acetonitrile (1 : 8 : 1, v/v/v) and a mixture of 50 mmol/L ammonium acetate and acetonitrile (1 : 9, v/v), respectively. The ratio of mobile phase B was increased linearly from 5 to 95% over 3 min, and then maintained at 95% over the next 1 min. Mobile phases A and B under acidic conditions were a mixture of 0.2% formic acid in 10 mmol/L ammonium formate and 0.2% formic acid in acetonitrile, respectively. The ratio of mobile phase B was increased linearly from 14 to 86% over 3 min, and then maintained at 86% over the next 1 min. All final test compounds were purified to >95% chemical purity as measured by analytical HPLC. Elemental analyses were carried out by Takeda Analytical Laboratories, and all results were within ±0.4% of the theoretical values. Melting points were determined on a BÜCHI B-545 melting point apparatus or a DSC1 system (Mettler-Toledo International Inc., Greifensee, Switzerland). Proton nuclear magnetic resonance (1H-NMR) spectra were recorded on a Varian Mercury-300 (300 MHz) or Bruker DPX300 (300 MHz) instrument. All 1H-NMR spectra were consistent with the proposed structures. All proton shifts are given in parts per million (ppm) downfield from tetramethysilane (δ) as the internal standard in deuterated solvent, and coupling constants (J) are in hertz (Hz). NMR data are reported as follows: chemical shift, integration, multiplicity (s, singlet; d, doublet; t, triplet; q, quartet; quint, quintet; m, multiplet; dd, doublet of doublets; td, triplet of doublets; ddd, doublet of doublet of doublets; and brs, broad singlet), and coupling constants. Very broad peaks for protons of, for example, hydroxyl and amino groups are not always indicated.
N-((1S)-2-Hydroxy-2-methyl-1-(4-(trifluoromethoxy)phenyl)propyl)-5-isopropylpyrazolo[1,5-a]pyrimidine-3-carboxamide (8)A mixture of 34 (38.6 mg, 0.09 mmol) and 5% Pd/C–ethylenediamine complex (10 mg) in MeOH (5 mL) was hydrogenated under balloon pressure at room temperature (r.t.) for 16 h. The catalyst was removed by filtration and the filtrate was concentrated in vacuo. The residue was purified by column chromatography (silica gel, hexane–ethyl acetate, 19 : 1 to 0 : 100) to afford 8 (32.1 mg, 0.074 mmol, 83%) as a white amorphous solid. 1H-NMR (300 MHz, DMSO-d6) δ: 0.99–1.08 (3H, m), 1.27 (3H, s), 1.41 (3H, d, J=4.5 Hz), 1.43 (3H, d, J=4.5 Hz), 3.20–3.34 (1H, m), 4.81–5.00 (2H, m), 7.22–7.34 (3H, m), 7.48–7.61 (2H, m), 8.44 (1H, s), 9.07 (1H, d, J=8.7 Hz), 9.19 (1H, d, J=7.2 Hz). MS (ESI/APCI) m/z 437.1 [M+H]+. HPLC purity: 98.9%.
N-((1S)-2-Hydroxy-2-methyl-1-(4-(trifluoromethoxy)phenyl)propyl)-5-(tetrahydro-2H-pyran-4-yl)pyrazolo[1,5-a]pyrimidine-3-carboxamide (9)A mixture of 35 (84.4 mg, 0.177 mmol) and Pd/C (en) (15 mg) in MeOH (10 mL) and tetrahydrofuran (THF) (dry) (2 mL) was hydrogenated under balloon pressure at r.t. for 16 h. The catalyst was removed by filtration and the filtrate was concentrated in vacuo. The residue was purified by column chromatography (silica gel, hexane–ethyl acetate, 19 : 1 to 0 : 100) to afford 9 (82.9 mg, 0.173 mmol, 98%) as a white amorphous solid. 1H-NMR (300 MHz, DMSO-d6) δ: 1.04 (3H, s), 1.28 (3H, s), 1.86–2.11 (4H, m), 3.13–3.28 (1H, m), 3.45–3.59 (2H, m), 3.97–4.10 (2H, m), 4.88–4.99 (2H, m), 7.24–7.34 (3H, m), 7.50–7.61 (2H, m), 8.45 (1H, s), 9.03 (1H, d, J=8.7 Hz), 9.21 (1H, d, J=7.2 Hz). MS (ESI/APCI) m/z 479.1 [M+H]+. HPLC purity: 99.6%.
N-((1S)-2-Hydroxy-2-methyl-1-(4-(trifluoromethoxy)phenyl)propyl)-5-(pyridin-2-yl)pyrazolo[1,5-a]pyrimidine-3-carboxamide (10)A mixture of crude 39 (79.0 mg, 0.329 mmol), 28 (94.0 mg, 0.329 mmol), HATU (163 mg, 0.428 mmol), N,N-diisopropylethylamine (DIEA) (0.172 mL, 0.987 mmol), and N,N-dimethylformamide (DMF) (3 mL) was stirred at r.t. for 12 h. The mixture was diluted with water and extracted with EtOAc. The organic layer was separated, washed with saturated aqueous NaCl, dried over anhydrous Na2SO4 and concentrated in vacuo. The residue was purified by column chromatography (basic silica gel, hexane–ethyl acetate, 100 : 0 to 0 : 100) to give 10 (130 mg, 0.276 mmol, 84% (63% yield in 2 steps from 39)) as a pale yellow amorphous solid. 1H-NMR (300 MHz, CDCl3) δ: 1.21 (3H, s), 1.49 (3H, s), 1.96 (1H, s), 5.14 (1H, d, J=8.3 Hz), 7.14–7.22 (2H, m), 7.44–7.56 (3H, m), 7.88–7.97 (1H, m), 8.26 (1H, d, J=7.4 Hz), 8.64 (1H, s), 8.75–8.88 (3H, m), 9.25 (1H, d, J=8.3 Hz). MS (ESI/APCI) m/z 472.1 [M+H]+. HPLC purity: 97.3%. Anal. Calcd for C23H20F3N5O3: C, 58.60; H, 4.28; N, 14.86. Found: C, 58.61; H, 4.51; N, 14.86.
N-((1S)-2-Hydroxy-2-methyl-1-(4-(trifluoromethoxy)phenyl)propyl)-5-(6-methylpyridin-3-yl)pyrazolo[1,5-a]pyrimidine-3-carboxamide (11)The title compound was prepared as a pale yellow solid after trituration with hexane–ethyl acetate (5 : 1) in 63% yield from 33 and 2-methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridine using the procedure analogous to that described for the synthesis of 35, except that basic silica gel was employed in a column chromatography purification in place of silica gel. 1H-NMR (300 MHz, DMSO-d6) δ: 1.03 (3H, s), 1.33 (3H, s), 2.60 (3H, s), 4.93 (1H, d, J=8.3 Hz), 5.19 (1H, s), 7.23–7.33 (2H, m), 7.48–7.59 (3H, m), 7.98 (1H, d, J=7.5 Hz), 8.50 (1H, s), 8.81 (1H, dd, J=8.3, 2.3 Hz), 9.24 (1H, d, J=8.7 Hz), 9.41 (1H, d, J=7.5 Hz), 9.54 (1H, d, J=2.3 Hz). MS (ESI/APCI) m/z 486.1 [M+H]+. HPLC purity: 100%. mp 237°C.
N-((1S)-2-Hydroxy-2-methyl-1-(4-(trifluoromethoxy)phenyl)propyl)-5-(2-methylpyridin-4-yl)pyrazolo[1,5-a]pyrimidine-3-carboxamide (12)A mixture of 33 (130 mg, 0.303 mmol), 2-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridine (100 mg, 0.455 mmol), (Amphos)2PdCl2 (21.5 mg, 0.0303 mmol), K2CO3 (62.9 mg, 0.455 mmol), toluene (2 mL) and water (0.2 mL) was heated at 120°C under microwave irradiation for 2.5 h. After being cooled to r.t., the reaction mixture was directly purified by column chromatography (basic silica gel, ethyl acetate–methanol, 100 : 0 to 19 : 1) to give 12 (115 mg, 0.237 mmol, 78%) as an off-white solid after recrystallization from heptane–ethyl acetate. 1H-NMR (300 MHz, CDCl3) δ: 1.21 (3H, s), 1.50 (3H, s), 1.73 (1H, s), 2.72 (3H, s), 5.13 (1H, d, J=8.5 Hz), 7.13–7.20 (2H, m), 7.46–7.54 (3H, m), 7.91 (1H, dd, J=5.1, 1.3 Hz), 8.09 (1H, s), 8.67 (1H, s), 8.75 (1H, d, J=5.3 Hz), 8.88 (1H, d, J=7.4 Hz), 9.19 (1H, d, J=8.3 Hz). MS (ESI/APCI) m/z 486.1 [M+H]+. HPLC purity: 100%. mp 192°C. Anal. Calcd for C24H22 F3N5O3: C, 59.38; H, 4.57; N, 14.43. Found: C, 59.03; H, 4.60; N, 14.24.
5-(1-(Difluoromethyl)-1H-pyrazol-4-yl)-N-((1S)-2-hydroxy-2-methyl-1-(4-(trifluoromethoxy)phenyl)propyl)pyrazolo[1,5-a]pyrimidine-3-carboxamide (13)The title compound was prepared as a white amorphous solid in 77% yield from 33 and 1-(difluoromethyl)-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole using the procedure analogous to that described for the synthesis of 35, except that the crude material was purified by basic silica gel column chromatography followed by silica gel column chromatography. 1H-NMR (300 MHz, DMSO-d6) δ: 1.04 (3H, s), 1.36 (3H, s), 4.89 (1H, d, J=8.3 Hz), 5.28 (1H, s), 7.23–7.35 (2H, m), 7.49–7.59 (2H, m), 7.73 (1H, d, J=7.5 Hz), 7.97 (1H, t, J=59.0 Hz), 8.45 (1H, s), 8.81 (1H, s), 9.29 (1H, s), 9.32–9.42 (2H, m). MS (ESI/APCI) m/z 511.1 [M+H]+. HPLC purity: 100%.
N-((1S)-2-Hydroxy-2-methyl-1-(4-(trifluoromethoxy)phenyl)propyl)-5-(1H-pyrazol-1-yl)pyrazolo[1,5-a]pyrimidine-3-carboxamide (14)To a mixture of 33 (126.3 mg, 0.295 mmol) and 1H-pyrazole (24.1 mg, 0.353 mmol) in DMF (4 mL) was added K2CO3 (48.8 mg, 0.353 mmol). The mixture was stirred at 90°C for 30 min and then poured into water. The mixture was extracted with EtOAc, washed with water and saturated aqueous NaCl, dried over anhydrous Na2SO4 and concentrated in vacuo. The residue was purified by column chromatography (silica gel, hexane–ethyl acetate, 19 : 1 to 0 : 100) to afford 14 (102 mg, 0.222 mmol, 75%) as a white solid after trituration with hexane–ethyl acetate (5 : 1). 1H-NMR (300 MHz, DMSO-d6) δ: 1.02 (3H, s), 1.36 (3H, s), 4.86 (1H, d, J=8.3 Hz), 5.34 (1H, s), 6.75–6.81 (1H, m), 7.23–7.33 (2H, m), 7.48–7.58 (2H, m), 7.76 (1H, d, J=7.5 Hz), 8.05 (1H, d, J=1.5 Hz), 8.46 (1H, s), 9.02–9.14 (2H, m), 9.37 (1H, d, J=7.5 Hz). MS (ESI/APCI) m/z 461.1 [M+H]+. HPLC purity: 100%. mp 241°C.
N-((1S)-2-Hydroxy-2-methyl-1-(4-(trifluoromethoxy)phenyl)propyl)-5-(4-methyl-1H-imidazol-1-yl)pyrazolo[1,5-a]pyrimidine-3-carboxamide (15)The title compound was prepared as a pale yellow solid after trituration with hexane–ethyl acetate (5 : 1) in 70% yield from 33 and 4-methyl-1H-imidazole using the procedure analogous to that described for the synthesis of 14, except that a preparative HPLC (column: L-Column2 ODS 20 mm ID×150 mm L; mobile phase A: 0.1% TFA in water; mobile phase B: 0.1% TFA in acetonitrile; flow rate: 20 mL/min) was employed in place of silica gel column chromatography. 1H-NMR (300 MHz, DMSO-d6) δ: 1.02 (3H, s), 1.35 (3H, s), 2.24 (3H, s), 4.87 (1H, d, J=8.3 Hz), 5.31 (1H, s), 7.24–7.33 (2H, m), 7.47–7.57 (2H, m), 7.73 (1H, d, J=7.9 Hz), 8.07 (1H, s), 8.45 (1H, s), 8.85 (1H, d, J=0.8 Hz), 9.00 (1H, d, J=8.3 Hz), 9.46 (1H, d, J=7.5 Hz). MS (ESI/APCI) m/z 475.1 [M+H]+. HPLC purity: 99.2%. mp 130°C.
(S)-N-(2-Hydroxy-2-methyl-1-(4-(trifluoromethoxy)phenyl)propyl)-5-(4-(trifluoromethyl)-1H-imidazol-1-yl)pyrazolo[1,5-a]pyrimidine-3-carboxamide (16)The title compound was prepared as a pale yellow solid after trituration with hexane–ethyl acetate (5 : 1) in 70% yield from 33 and 4-(trifluoromethyl)-1H-imidazole using the procedure analogous to that described for the synthesis of 14. 1H-NMR (300 MHz, DMSO-d6) δ: 1.02 (3H, s), 1.34 (3H, s), 4.86 (1H, d, J=8.3 Hz), 5.36 (1H, s), 7.23–7.33 (2H, m), 7.47–7.58 (2H, m), 7.89 (1H, d, J=7.5 Hz), 8.53 (1H, s), 8.92–9.02 (2H, m), 9.15 (1H, s), 9.62 (1H, d, J=7.5 Hz). MS (ESI/APCI) m/z 529.1 [M+H]+. HPLC purity: 100%. mp 220°C.
N-((1S)-2-Hydroxy-2-methyl-1-(4-(trifluoromethoxy)phenyl)propyl)-5-(4-methyl-1H-1,2,3-triazol-1-yl)pyrazolo[1,5-a]pyrimidine-3-carboxamide (17)A mixture of 33 (430 mg, 1.00 mmol), 4-methyl-1H-1,2,3-triazole (100 mg, 1.20 mmol) and K2CO3 (208 mg, 1.50 mmol) in DMF (4 mL) was stirred at 80°C for 2 h. The mixture was quenched with water at r.t. and extracted with EtOAc. The organic layer was separated, washed with water and saturated aqueous NaCl, dried over anhydrous MgSO4 and concentrated in vacuo (508.7 mg). The residue was purified by using preparative HPLC (column: L-Column2 ODS 20 mm ID×150 mm L; mobile phase A: 0.1% TFA in water; mobile phase B: 0.1% TFA in acetonitrile; flow rate: 20 mL/min). The first eluting factions (tR1) were concentrated to dryness, and washed with saturated aqueous NaHCO3, extracted with EtOAc. The organic layer was separated, washed with water and saturated aqueous NaCl, dried over anhydrous MgSO4 and concentrated in vacuo. The residue (42.7 mg) was crystallized from hexane–ethyl acetate to give 17 (34.4 mg, 0.072 mmol, 7.2%) as a white solid. 1H-NMR (300 MHz, DMSO-d6) δ: 1.04 (3H, s), 1.37 (3H, s), 2.44 (3H, s), 4.87 (1H, d, J=8.3 Hz), 5.39 (1H, s), 7.23–7.34 (2H, m), 7.48–7.58 (2H, m), 7.93 (1H, d, J=7.5 Hz), 8.55 (1H, s), 8.88 (1H, d, J=0.8 Hz), 9.07 (1H, d, J=8.3 Hz), 9.50 (1H, d, J=7.5 Hz). MS (ESI/APCI) m/z 476.2 [M+H]+. HPLC purity: 99.4%. mp 260°C.
N-((1S)-2-Hydroxy-2-methyl-1-(4-(trifluoromethoxy)phenyl)propyl)-5-(3-methyl-1H-1,2,4-triazol-1-yl)pyrazolo[1,5-a]pyrimidine-3-carboxamide (18)The title compound was prepared as a white solid after trituration with diisopropyl ether in 35% yield from 33 and 3-methyl-1H-1,2,4-triazole using the procedure analogous to that described for the synthesis of 14, except that basic silica gel was employed in a column chromatography purification in place of silica gel. 1H-NMR (300 MHz, DMSO-d6) δ: 1.02 (3H, br s), 1.36 (3H, s), 2.47 (3H, br s), 4.86 (1H, d, J=8.1 Hz), 5.43 (1H, s), 7.21–7.35 (2H, m), 7.47–7.59 (2H, m), 7.63 (1H, d, J=7.5 Hz), 8.52 (1H, s), 9.06 (1H, d, J=8.1 Hz), 9.46 (1H, d, J=7.5 Hz), 9.55 (1H, s). MS (ESI/APCI) m/z 476.1 [M+H]+. HPLC purity: 100%.
(S)-N-(2-Hydroxy-2-methyl-1-(4-(trifluoromethoxy)phenyl)propyl)-6-methyl-5-(4-methyl-1H-1,2,3-triazol-1-yl)pyrazolo[1,5-a]pyrimidine-3-carboxamide (19)To a mixture of 52a (2.81 g, 10.9 mmol) and 28 (3.26 g, 11.4 mmol) in DMF (250 mL) were added HOBt·H2O (2.06 g, 13.1 mmol), EDCI·HCl (2.55 g, 13.1 mmol) and triethylammonium acetate (TEA) (7.65 mL, 54.4 mmol). The mixture was stirred at r.t. overnight and then quenched with water at r.t. The mixture was extracted with EtOAc, washed with saturated aqueous NaHCO3, water, and then saturated aqueous NaCl, dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was purified by column chromatography (silica gel, hexane–ethyl acetate, 100 : 0 to 3 : 17) to give 19 (3.86 g, 7.89 mmol, 73%) as a white solid. 1H-NMR (300 MHz, CDCl3) δ: 1.17 (3H, s), 1.48 (3H, s), 1.74 (1H, s), 2.50 (3H, d, J=0.8 Hz), 2.84 (3H, d, J=0.8 Hz), 5.05(1H, d, J=8.3 Hz), 7.17 (2H, d, J=7.9 Hz), 7.38–7.48 (2H, m), 8.60 (1H, s), 8.69–8.82 (3H, m). The obtained product (3.86 g, 7.89 mmol) was dissolved in EtOAc (277 mL) at 50–60°C. To this solution was added hexane (150 mL) at the same temperature and a white solid precipitated. After 2 h of stirring at r.t., further hexane (88 mL) was added to the mixture, which was stirred at r.t. for 10 min. The white precipitate was collected by filtration, washed with hexane–ethyl acetate, and dried to give 19 (3.17 g, 6.48 mmol, 82%) as a white solid. 1H-NMR (300 MHz, CDCl3) δ: 1.17 (3H, s), 1.48 (3H, s), 1.74 (1H, s), 2.50 (3H, d, J=0.8 Hz), 2.84 (3H, d, J=0.8 Hz), 5.05 (1H, d, J=8.3 Hz), 7.17 (2H, d, J=8.3 Hz), 7.39–7.48 (2H, m), 8.60 (1H, s), 8.69–8.83 (3H, m). MS (ESI/APCI) m/z 490.2 [M+H]+. HPLC purity: 100%. mp 238–239°C. Anal. Calcd for C22H22F3N7O3: C, 53.99; H, 4.53; F, 11.64; N, 20.03. Found: C, 53.82; H, 4.42; F, 11.64; N, 19.81.
(S)-N-(2-Hydroxy-2-methyl-1-(4-(trifluoromethoxy)phenyl)propyl)-6-methyl-5-(3-methyl-1H-1,2,4-triazol-1-yl)pyrazolo[1,5-a]pyrimidine-3-carboxamide (20)To a suspension of 52b (606 mg, 2.35 mmol) in DMF (12 mL) were added 28 (805 mg, 2.82 mmol), EDCI·HCl (540 mg, 2.82 mmol), HOBt·H2O (431 mg, 2.82 mmol), and TEA (0.392 mL, 2.82 mmol) at r.t. The mixture was stirred at r.t. for 2 h. The mixture was poured into water and extracted with EtOAc. The organic layer was separated, washed with saturated aqueous NaCl, dried over anhydrous MgSO4 and concentrated in vacuo. The residue was purified by column chromatography (basic silica gel, hexane–ethyl acetate, 4 : 1 to 1 : 9) to give 20 (858 mg, 1.75 mmol, 75%) as a white solid after recrystallization from diisopropyl ether/ethyl acetate. 1H-NMR (300 MHz, DMSO-d6) δ: 1.00 (3H, s), 1.31 (3H, s), 2.46 (3H, s), 2.64 (3H, d, J=0.8 Hz), 4.87 (1H, d, J=8.0 Hz), 5.32 (1H, s), 7.27 (2H, d, J=8.0 Hz), 7.51 (2H, d, J=8.7 Hz), 8.49 (1H, s), 8.85 (1H, d, J=8.3 Hz), 9.46–9.53 (2H, m). MS (ESI/APCI) m/z 490.3 [M+H]+. HPLC purity: 100%. mp 212°C. Anal. Calcd for C22H22F3N7O3: C, 53.99; H, 4.53; N, 20.03. Found: C, 54.11; H, 4.50; N, 20.04.
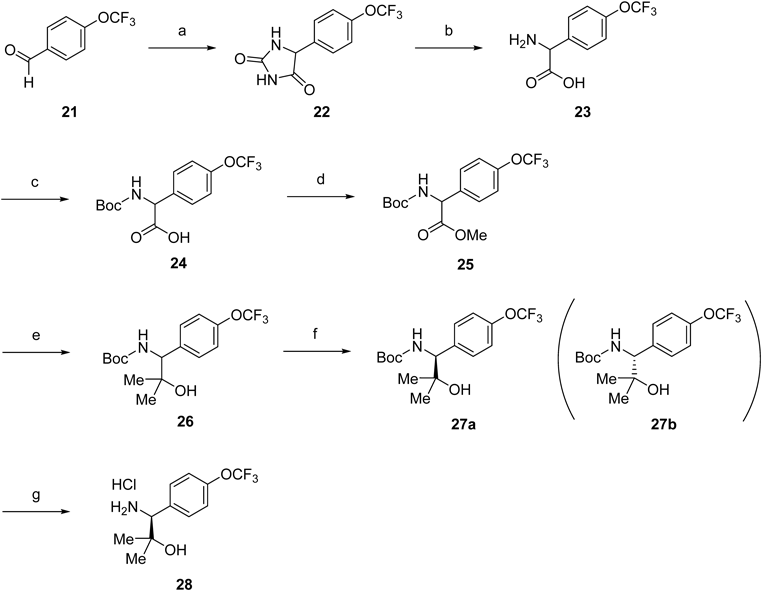
2-Amino-2-(4-(trifluoromethoxy)phenyl)acetic Acid (23)The solution of potassium cyanide (19.4 g, 299 mmol) in water (170 mL) at 50°C was added dropwise to a solution of 4-trifluoromethoxy benzaldehyde (21) (34.1 mL, 239 mmol) and ammonium carbonate (62.0 g, 645 mmol) in EtOH (273 mL) and water (109 mL). The mixture was stirred at 60°C for 3 h. After cooling to r.t., EtOH was removed under reduced pressure. To the mixture was acidified to pH ca. 1 with concd HCl at 0°C. The solid was filtered off and washed with water. To a solution of potassium hydroxide (66.2 g, 1000 mmol) in water (250 mL) was added the product at r.t. The mixture was stirred at 90°C overweekend. After cooling to r.t., the mixture was neutralized with concd HCl. The resulting solid was filtered off, washed with water, and dried to give 23 (56.0 g, 238 mmol, 100%) as a pale yellow solid. This was used in the next reaction without further purification. MS (ESI/APCI) m/z 234.0 [M−H]−.
((tert-Butoxycarbonyl)amino)(4-(trifluoromethoxy)phenyl)acetic Acid (24)To a solution of 23 (56.0 g, 238 mmol) in THF (476 mL) was added Boc2O (82.9 mL, 357 mmol) and 2 M NaOH aqueous solution (357 mL, 714 mmol) at r.t. The mixture was stirred at r.t. The mixture was poured into water at r.t. and extracted with Et2O. The aqueous layer was acidified to pH ca. 3 with 1 M HCl aqueous solution at 0°C and then extracted with EtOAc. The organic layer was washed with saturated aqueous NaCl, dried over anhydrous MgSO4 and concentrated in vacuo to afford 24 (63.3 g, 189 mmol) as a pale yellow solid. This was subjected to the next reaction without further purification. MS (ESI/APCI) m/z 333.9 [M−H]−.
tert-Butyl (2-Hydroxy-2-methyl-1-(4-(trifluoromethoxy)phenyl)propyl)carbamate (25)To a solution of 24 (63.3 g, 189 mmol) in DMF (378 mL) were added MeI (14.2 mL, 227 mmol) and K2CO3 (31.3 g, 227 mmol) at r.t. The mixture was stirred at r.t. for 2 h. The mixture was poured into water at r.t. and extracted with EtOAc. The organic layer was separated, washed with saturated aqueous NaCl, dried over anhydrous MgSO4 and concentrated in vacuo. The residue was purified by column chromatography (silica gel, hexane–ethyl acetate, 100 : 0 to 4 : 1) to give 25 (38.7 g, 111 mmol, 46% from 21) as an off-white solid. 1H-NMR (300 MHz, CDCl3) δ: 1.26–1.53 (9H, m), 3.73 (3H, s), 5.27–5.41 (1H, m), 5.54–5.75 (1H, m), 7.20 (2H, d, J=8.3 Hz), 7.35–7.44 (2H, m). MS (ESI/APCI) m/z 348.1 [M−H]−.
tert-Butyl (2-Hydroxy-2-methyl-1-(4-(trifluoromethoxy)phenyl)propyl)carbamate (26)To a solution of 25 (23.5 g, 67.3 mmol) in THF (336 mL) was added dropwise MeMgBr (269 mL, 269 mmol) at 0°C. The mixture was stirred at 0°C under Ar for 1 h. The mixture was quenched with saturated aqueous NH4Cl at 0°C and extracted with EtOAc. The organic layer was separated, washed with saturated aqueous NaCl, dried over anhydrous MgSO4 and concentrated in vacuo. The residue was purified by column chromatography (silica gel, hexane–ethyl acetate, 19 : 1 to 2 : 3) to give 26 (17.9 g, 51.2 mmol, 76%) as a white solid. 1H-NMR (300 MHz, CDCl3) δ: 1.05 (3H, s), 1.30–1.47 (12H, m), 4.50 (1H, d, J=6.4 Hz), 5.53 (1H, d, J=8.7 Hz), 7.18 (2H, d, J=7.9 Hz), 7.33 (2H, d, J=8.7 Hz). MS (ESI/APCI) m/z 348.0 [M−H]−.
tert-Butyl ((1S)-2-Hydroxy-2-methyl-1-(4-(trifluoromethoxy)phenyl)propyl)carbamate (27a)Resolution of the enantiomers of 26 was carried out chromatographically using a Chiralpak AD 20 mm ID×250 mm L column (hexane–ethanol, 950 : 50) at 80 mL/min. Resolution of 26 (22.3 g, 63.8 mmol) provided 27a as a white solid (10.2 g, 29.1 mmol, 46%, 91% theoretical) as the first eluting enantiomer. Analytical HPLC analysis carried out on a 4.6 mm ID×250 mm L Chiralpak AD column with the same eluent as above at a flow rate of 1.0 mL/min indicated that 27a was of >99.9% ee. 1H-NMR (300 MHz, CDCl3) δ: 1.05 (3H, s), 1.36 (3H, s), 1.40 (9H, br s), 1.51 (1H, br s), 4.50 (1H, d, J=7.5 Hz), 5.53 (1H, d, J=8.3 Hz), 7.13–7.23 (2H, m), 7.29–7.39 (2H, m).
(1S)-1-Amino-2-methyl-1-(4-(trifluoromethoxy)phenyl)propan-2-ol Hydrochloride (28)A mixture of 27a (10.2 g, 29.3 mmol) and 4 M HCl solution in EtOAc (70.0 mL, 280 mmol) was stirred at r.t. for 1.5 h. The mixture was concentrated in vacuo and the resulting solid was triturated with hexane–diisopropyl ether, collected by filtration, rinsed with hexane–diisopropyl ether, and dried to afford 28 (7.00 g, 24.5 mmol, 84%) as a pale red solid. 1H-NMR (300 MHz, DMSO-d6) δ: 0.98 (3H, s), 1.23 (3H, s), 4.23 (1H, br s), 5.39 (1H, s), 7.37–7.48 (2H, m), 7.63 (2H, d, J=8.7 Hz), 8.46 (3H, br s).
Ethyl 5-Hydroxypyrazolo[1,5-a]pyrimidine-3-carboxylate (30)To a mixture of ethyl 3-amino-1H-pyrazole-4-carboxylate (29) (30.0 g, 193 mmol) and ethyl 3-ethoxy-2-propenoate (cis- and trans-mixture, 41.9 mL, 290 mmol) in DMF (387 mL) was added cesium carbonate (113 g, 348 mmol). The mixture was stirred at 100°C for 2 h, diluted with water and then acidified to pH ca. 5 with AcOH. The resulting solid was filtered by filtration, washed with water and dried to afford 30 (36.4 g, 176 mmol, 91%) as a beige solid. 1H-NMR (300 MHz, DMSO-d6) δ: 1.28 (3H, t, J=7.1 Hz), 4.28 (2H, q, J=7.1 Hz), 6.15 (1H, d, J=7.9 Hz), 8.13 (1H, s), 8.57 (1H, d, J=7.9 Hz), 11.73 (1H, br s).
5-Hydroxypyrazolo[1,5-a]pyrimidine-3-carboxylic Acid (31)To a solution of 30 (20.8 g, 100 mmol) in THF (223 mL) and EtOH (111 mL) was added 2 M NaOH aqueous solution (201 mL, 401 mmol). The mixture was stirred at 50°C overnight. The mixture was evaporated under reduced pressure to remove solvents and then neutralized with 2 M HCl aqueous solution (201 mL, 401 mmol). The resulting solid was collected by filtration, rinsed with water/EtOH and dried to give 31 (17.7 g, 99 mmol, 98%) as a beige solid. 1H-NMR (300 MHz, DMSO-d6) δ: 6.14 (1H, d, J=7.9 Hz), 8.09 (1H, s), 8.56 (1H, d, J=7.9 Hz).
5-Chloropyrazolo[1,5-a]pyrimidine-3-carbonyl Chloride (32)To cooled (0°C) POCl3 (71.0 mL, 762 mmol) were added 31 (5.00 g, 27.9 mmol) and DIEA (16.1 mL, 92.1 mmol). The mixture was stirred at 130°C for 4 h and then concentrated in vacuo. The residue was diluted with toluene and water at 0°C, and then extracted with EtOAc. The organic layer was separated, dried over anhydrous Na2SO4, filtered through a cake of silica gel pad (eluted with ethyl acetate) and concentrated in vacuo. The resulting solid was triturated with heptane, collected by filtration, washed with heptane and dried to give 32 (4.46 g, 20.7 mmol, 74%) as a beige solid. 1H-NMR (300 MHz, CDCl3) δ: 7.16 (1H, d, J=7.2 Hz), 8.65 (1H, s), 8.70 (1H, d, J=7.2 Hz).
5-Chloro-N-((1S)-2-hydroxy-2-methyl-1-(4-(trifluoromethoxy)phenyl)propyl)pyrazolo[1,5-a]pyrimidine-3-carboxamide (33)To a cooled (0°C) mixture of 32 (1.48 g, 6.85 mmol) and CH3CN (30 mL) were added 28 (1.96 g, 6.85 mmol) and DIEA (3.58 mL, 20.6 mmol), and the mixture was stirred at 0°C to r.t. for 16 h. The mixture was poured into water and extracted with EtOAc. The organic layer was separated, washed with saturated aqueous NaCl, dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was purified by column chromatography (silica gel, hexane–ethyl acetate, 100 : 0 to 0 : 100) to give 33 (2.27 g, 5.29 mmol, 77%) as white amorphous solids. 1H-NMR (300 MHz, CDCl3) δ: 1.19 (3H, s), 1.40 (3H, s), 1.88 (1H, s), 5.12 (1H, d, J=8.7 Hz), 6.99 (1H, d, J=7.2 Hz), 7.17–7.23 (2H, m), 7.45–7.53 (2H, m), 8.56 (1H, d, J=8.7 Hz), 8.62 (1H, s), 8.67 (1H, d, J=7.2 Hz). MS (ESI/APCI) m/z 429.1 [M+H]+.
N-((1S)-2-Hydroxy-2-methyl-1-(4-(trifluoromethoxy)phenyl)propyl)-5-(prop-1-en-2-yl)pyrazolo[1,5-a]pyrimidine-3-carboxamide (34)Into a microwave vial equipped with a magnetic stirrer were added 33 (76.1 mg, 0.177 mmol), 4,4,5,5-tetramethyl-2-(prop-1-en-2-yl)-1,3,2-dioxaborolane (46.6 mg, 0.266 mmol), K2CO3 (36.8 mg, 0.266 mmol), toluene (4 mL) and water (0.4 mL), followed by (Amphos)2PdCl2 (12.6 mg, 0.0177 mmol). The reaction vial was flushed with nitrogen, sealed and heated by microwave irradiation at 150°C for 25 min. The mixture was purified by column chromatography (basic silica gel, hexane–ethyl acetate, 19 : 1 to 1 : 4), followed by a second column purification (silica gel, hexane–ethyl acetate, 19 : 1 to 1 : 4) to afford 34 (48.8 mg, 0.112 mmol, 63%) as a yellow amorphous solid. 1H-NMR (300 MHz, DMSO-d6) δ: 1.00 (3H, s), 1.27 (3H, s), 2.37 (3H, s), 4.88–4.98 (2H, m), 5.80 (1H, s), 6.36 (1H, s), 7.24–7.34 (2H, m), 7.49–7.58 (2H, m), 7.65 (1H, d, J=7.5 Hz), 8.45 (1H, s), 9.02 (1H, d, J=8.7 Hz), 9.22 (1H, d, J=7.5 Hz). MS (ESI/APCI) m/z 435.2 [M+H]+.
5-(3,6-Dihydro-2H-pyran-4-yl)-N-((1S)-2-hydroxy-2-methyl-1-(4-(trifluoromethoxy)phenyl)propyl)pyrazolo[1,5-a]pyrimidine-3-carboxamide (35)Into a microwave vial equipped with a magnetic stirrer were added 33 (142 mg, 0.330 mmol), 2-(3,6-dihydro-2H-pyran-4-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (104 mg, 0.495 mmol), K2CO3 (68.5 mg, 0.495 mmol), toluene (4 mL) and water (0.4 mL), followed by (Amphos)2PdCl2 (23.4 mg, 0.0330 mmol). The reaction vial was flushed with nitrogen, sealed, and heated by microwave irradiation at 150°C for 25 min. The mixture was purified by column chromatography (silica gel, hexane–ethyl acetate, 19 : 1 to 1 : 4) to afford 35 (110 mg, 0.230 mmol, 70%) as a yellow solid. 1H-NMR (300 MHz, DMSO-d6) δ: 1.00 (3H, s), 1.28 (3H, s), 2.67–2.94 (2H, m), 3.84–3.97 (2H, m), 4.35–4.43 (2H, m), 4.86–5.00 (2H, m), 7.24–7.34 (3H, m), 7.49–7.56 (2H, m), 7.59 (1H, d, J=7.5 Hz), 8.42 (1H, s), 9.06 (1H, d, J=8.7 Hz), 9.20 (1H, d, J=7.5 Hz). MS (ESI/APCI) m/z 477.2 [M+H]+.
Ethyl 5-Chloropyrazolo[1,5-a]pyrimidine-3-carboxylate (36)POCl3 (30 mL, 322 mmol) was added to 30 (5.54 g, 26.7 mmol) and the mixture was stirred at 100°C for 16 h. After POCl3 was removed under reduced pressure, the residue was partitioned between EtOAc and NaHCO3 aqueous solution. The phases were separated and the aqueous phase was extracted with EtOAc. The combined organic phases were washed with water and saturated aqueous NaCl, dried over anhydrous Na2SO4 and concentrated in vacuo. The residue was purified by column chromatography (silica gel, hexane–ethyl acetate, 19 : 1 to 1 : 1) to afford 36 (3.69 g, 16.3 mmol, 61%) as a white solid. MS (ESI/APCI) m/z 226.1 [M+H]+.
Ethyl 5-(Pyridin-2-yl)pyrazolo[1,5-a]pyrimidine-3-carboxylate (38)A mixture of ethyl 36 (300 mg, 1.33 mmol), lithium 4-methyl-1-(pyridin-2-yl)-2,6,7-trioxa-1-borabicyclo[2.2.2]octan-1-uide (37) (566 mg, 2.66 mmol), Ph3P (139 mg, 0.532 mmol), copper(I) iodide (127 mg, 0.665 mmol) and Pd(OAc)2 (29.9 mg, 0.133 mmol) in DMF (5 mL) was stirred at 80°C for 2 h under Ar. The mixture was filtered through a cake of basic silica gel pad (eluted with ethyl acetate), followed by a cake of silica gel pad (eluted with ethyl acetate). The appropriate fractions were concentrated in vacuo. The resulting solid was triturated with hot ethyl acetate and insoluble materials were filtered off. After concentration of the filtrate, the residue was purified by column chromatography (basic silica gel, hexane–ethyl acetate, 100 : 0 to 3 : 7) to give 38 (309 mg, 1.15 mmol, 87%) as a pale yellow solid after trituration with diisopropyl ether. 1H-NMR (300 MHz, CDCl3) δ: 1.47 (3H, t, J=7.1 Hz), 4.46 (2H, q, J=7.1 Hz), 7.40–7.47 (1H, m), 7.86–7.95 (1H, m), 8.25 (1H, d, J=7.4 Hz), 8.59 (1H, s), 8.71–8.76 (2H, m), 8.80 (1H, d, J=7.4 Hz). MS (ESI/APCI) m/z 269.1 [M+H]+.
5-(Pyridin-2-yl)pyrazolo[1,5-a]pyrimidine-3-carboxylic Acid (39)A mixture of 38 (117 mg, 0.436 mmol), 4 M NaOH aqueous solution (0.545 mL, 2.18 mmol), THF (3.5 mL) and EtOH (3.5 mL) was stirred at 60°C for 2 h. After being cooled to 0°C, the mixture was acidified with 6 M HCl aqueous solution (0.363 mL) and concentrated in vacuo to give crude 39 (79.0 mg, 0.329 mmol, 75%). This was used in the next reaction without further purification.
tert-Butyl 2-Cyano-3-(dimethylamino)acrylate (42)A mixture of tert-butyl 2-cyanoacetate (40) (24.3 g, 172 mmol) and tert-butoxy bis(dimethylamino)methane (41) (35.5 mL, 172 mmol) was stirred at r.t. for 30 min. The mixture was evaporated to give 42 as a pale yellow solid. This was used in the next reaction without further purification. 1H-NMR (300 MHz, CDCl3) δ: 1.50 (9H, s), 3.17 (3H, s), 3.36 (3H, s), 7.62 (1H, s).
tert-Butyl 3-Amino-1H-pyrazole-4-carboxylate (43)A mixture of 42 (33.8 g, 172 mmol) and hydrazine monohydrate (8.39 mL, 172 mmol) in MeOH (344 mL) was stirred at 70°C overnight. After cooling to r.t., the mixture was poured into water at r.t. and extracted with EtOAc. The organic layer was separated, washed with saturated aqueous NaCl, dried over anhydrous MgSO4 and concentrated in vacuo. The residue was crystallized from diisopropyl ether to give 43 (20.0 g, 109 mmol, 64%) as a pale orange solid. 1H-NMR (300 MHz, DMSO-d6) δ: 1.47 (9H, s), 4.86–6.14 (2H, m), 7.07–7.92 (1H, m), 11.44–12.34 (1H, m). MS (ESI/APCI) m/z 184.1 [M+H]+.
Methyl 2,3-Dibromo-2-methylpropanoate (45)To a solution of methyl methacrylate (60.1 g, 600 mmol) in EtOAc (353 mL) at 0°C was added a solution of bromine (30.7 mL, 600 mmol) in EtOAc (30 mL). The mixture was stirred at r.t. for 16 h and then saturated aqueous Na2S2O3 was added. The mixture was diluted with water and extracted with EtOAc. The combined organic phases were washed with saturated aqueous NaCl, dried over anhydrous Na2SO4 and concentrated in vacuo to afford 45 (152 g, 584 mmol, 97%) as a colorless oil. 1H-NMR (300 MHz, DMSO-d6) δ: 1.95 (3H, s), 3.76 (3H, s), 4.08–4.14 (1H, m), 4.16–4.22 (1H, m).
Methyl 3,3-Dimethoxy-2-methylpropanoate (46)A suspension of sodium methoxide (28% MeOH solution, 226 g, 1.17 mol) in MeOH (234 mL) was heated to 70°C and then a solution of 45 (152 g, 584 mmol) in MeOH (20 mL) was added rapidly. The mixture was stirred at 70°C for 3 h. After cooling to r.t., the mixture was filtered and washed with MeOH, and then the filtrate was concentrated in vacuo. The residue was partitioned between Et2O and water. The organic layer was washed with water and saturated aqueous NaCl, dried over anhydrous MgSO4, filtered, and concentrated in vacuo to give 46 (54.5 g, 336 mmol, 58%) as a pale yellow oil. This was used in the next reaction without further purification. 1H-NMR (300 MHz, CDCl3) δ: 1.17 (3H, d, J=7.2 Hz), 2.71–2.85 (1H, m), 3.35 (3H, s), 3.38 (3H, s), 3.70 (3H, s), 4.50 (1H, d, J=7.6 Hz).
tert-Butyl 5-Hydroxy-6-methylpyrazolo[1,5-a]pyrimidine-3-carboxylate (47)To a mixture of 43 (20.0 g, 109 mmol) and 46 (26.6 g, 164 mmol) in DMF (219 mL) was added cesium carbonate (64.2 g, 197 mmol). The mixture was stirred at 100°C for 16 h, diluted with water and then acidified to pH ca. 4 with AcOH. The resulting solid was filtered by filtration, washed with water and dried to afford 47 (22.6 g, 91.8 mmol, 84%) as a white solid. 1H-NMR (300 MHz, DMSO-d6) δ: 1.53 (9H, s), 1.96 (3H, d, J=1.1 Hz), 7.97 (1H, s), 8.53 (1H, d, J=1.1 Hz), 11.22 (1H, br s). MS (ESI/APCI) m/z 250.1 [M+H]+.
tert-Butyl 5-Azido-6-methylpyrazolo[1,5-a]pyrimidine-3-carboxylate (48)To a suspension of 47 (4.99 g, 20.0 mmol) in THF (100 mL) was added 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) (4.52 mL, 30.0 mmol) and DPPA (5.17 mL, 24.0 mmol) at r.t. The mixture was stirred at 60°C under Ar for 3 h. DBU (1.51 mL, 10.0 mmol) and DPPA (1.72 mL, 8.00 mmol) were added thereto. The mixture was stirred at 60°C under Ar for 2 h. The mixture was poured into water at r.t. and extracted with a 1 : 1 mixture of hexane and EtOAc. The organic layer was separated, washed with saturated aqueous NaCl, dried over anhydrous MgSO4, filtered through a short pad of silica gel, eluted with a 1 : 1 mixture of hexane and EtOAc, and concentrated in vacuo. The resulting solid was triturated with hexane to give 48 (4.03 g, 14.7 mmol, 74%) as a pale green solid. 1H-NMR (300 MHz, DMSO-d6) δ: 1.51–1.65 (9H, m), 2.12–2.55 (3H, m), 8.38–8.49 (1H, m), 8.85–9.11 (1H, m). MS (ESI/APCI) m/z 275.1 [M+H]+.
tert-Butyl 6-Methyl-5-(4-methyl-1H-1,2,3-triazol-1-yl)pyrazolo[1,5-a]pyrimidine-3-carboxylate (51a)A mixture of 48 (4.36 g, 15.9 mmol) and 49 (6.07 g, 19.1 mmol) in toluene (79 mL) was stirred at 80°C for 3 h. To the mixture was added MgCl2 (4.54 g, 47.7 mmol). After 15 min, to the mixture was added MgCl2 (3.03 g, 31.8 mmol). The mixture was stirred at 60°C under Ar for 2 h. The solid was filtered off, washed with hot toluene. The filtrate was evaporated. The residue was purified by column chromatography (silica gel, hexane–ethyl acetate, 19 : 1 to 9 : 11) to give 51a (4.83 g, 15.4 mmol, 97%) as a white solid. 1H-NMR (300 MHz, CDCl3) δ: 1.64 (9H, s), 2.46 (3H, d, J=0.8 Hz), 2.78 (3H, d, J=1.1 Hz), 8.46–8.50 (2H, m), 8.71–8.73 (1H, m). MS (ESI/APCI) m/z 315.2 [M+H]+.
6-Methyl-5-(4-methyl-1H-1,2,3-triazol-1-yl)pyrazolo[1,5-a]pyrimidine-3-carboxylic Acid (52a)To a stirred suspension of 51a (2.64 g, 8.40 mmol) in CH3CN (84 mL) was added methanesulfonic acid (2.78 mL, 42.0 mmol) at 0°C. The mixture was stirred at r.t. overnight. The starting material remained on TLC and further methanesulfonic acid (0.555 mL, 8.40 mmol) was added. The mixture was stirred at r.t. for 1 h and then neutralized with 1 M NaOH aqueous solution at 0°C. The organic solvent was evaporated under reduced pressure and then diluted with water. The resulting solid was collected by filtration, washed with water and then hexane, and dried to give 52a (1.98 g, 7.67 mmol, 91%) as a white solid. 1H-NMR (300 MHz, DMSO-d6) δ: 2.40 (3H, s), 2.52 (3H, d, J=0.8 Hz), 8.50 (1H, d, J=1.1 Hz), 8.62 (1H, s), 9.50 (1H, d, J=1.1 Hz), 12.52 (1H, br s). MS (ESI/APCI) m/z 259.2 [M+H]+.
tert-Butyl 5-Chloro-6-methylpyrazolo[1,5-a]pyrimidine-3-carboxylate (50)Carbon tetrachloride (9.73 mL, 100 mmol) was added to a solution of PPh3 (27.1 g, 100 mmol) in 1,2-dichloroethane (223 mL) at r.t. The mixture was stirred at the same temperature under N2 for 30 min. To this mixture was added a suspension of 47 (5.0 g, 20.1 mmol) in 1,2-dichloroethane (111 mL) at r.t. and the resulting mixture was stirred under N2 at 75–85°C for 4.5 h, and then concentrated in vacuo. The residue was partitioned between EtOAc and water. The phases were separated and the aqueous phase was extracted with EtOAc. The combined organic phases were washed with saturated aqueous NaCl, dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was purified by column chromatography (silica gel, hexane–ethyl acetate, 100 : 0 to 7 : 3) to give 50 (4.75 g, 17.7 mmol, 88%) as a white solid. 1H-NMR (300 MHz, CDCl3) δ: 1.62 (9H, s), 2.42 (3H, d, J=0.9 Hz), 8.40 (1H, s), 8.52 (1H, d, J=0.9 Hz).
tert-Butyl 6-Methyl-5-(3-methyl-1H-1,2,4-triazol-1-yl)pyrazolo[1,5-a]pyrimidine-3-carboxylate (51b)To a solution of 50 (1.08 g, 4.03 mmol) in DMF (20 mL) was added 3-methyl-1H-1,2,4-triazole (0.469 g, 5.65 mmol) and K2CO3 (0.781 g, 5.65 mmol) at r.t. The mixture was stirred at r.t. overnight and warmed to 60°C for 2 h. After cooling to r.t., water was added to the mixture. The solid was collected by filtration, washed with water, and dried to give 51b (0.870 g, 2.77 mmol, 69%) as a tan solid. 1H-NMR (300 MHz, DMSO-d6) δ: 1.56 (9H, s), 2.43 (3H, s), 2.54 (3H, d, J=1.1 Hz), 8.56 (1H, s), 9.10 (1H, s), 9.43 (1H, d, J=1.1 Hz). MS (ESI/APCI) m/z 315.2 [M+H]+.
6-Methyl-5-(3-methyl-1H-1,2,4-triazol-1-yl)pyrazolo[1,5-a]pyrimidine-3-carboxylic Acid (52b)To a suspension of 51b (0.87 g, 2.77 mmol) in CH3CN (14 mL) was added MsOH (1.26 mL, 19.4 mmol) at 0°C. After being stirred at r.t. for 5 h, the mixture was warmed to 60°C for 30 min. One molar NaOH aqueous solution (19.4 mL, 19.4 mmol) was added thereto at 0°C. CH3CN was evaporated. The solid was collected by filtration, washed with water, dried to give 52b (0.606 g, 2.35 mmol, 85%) as a tan solid. 1H-NMR (300 MHz, DMSO-d6) δ: 2.42 (3H, s), 2.48–2.53 (3H, m), 8.60 (1H, s), 9.09 (1H, s), 9.44 (1H, d, J=1.1 Hz), 12.49 (1H, br s). MS (ESI/APCI) m/z 259.1 [M+H]+.
Enzyme Assay ProtocolPreparation of Human PDEHuman PDE1A, 3A, 4D2, 5A1, 7B, 8A1, 9A2, and 11A4 enzymes were purchased from BPS Bioscience. Human PDE6AB enzyme was purchased from SB Drug Discovery. Human PDE2A3 full-length gene was transduced into Sf9 cells, and the enzyme was purified by His-tag affinity column and gel filtration. Human PDE10A2 was generated from COS-7 cells that had been transfected with the full-length gene. The enzymes were stored at −70°C until use.
PDE2A3 Enzyme Inhibitory AssayPDE activity was measured using an SPA (Scintillation Proximity Assay) (GE Healthcare). To evaluate the inhibitory activity of a compound, 10 µL of serially diluted compounds were incubated with 20 µL of PDE enzyme (final concentration 0.023 nM) in assay buffer (50 mM N-(2-hydroxyethyl)piperazine-N′-2-ethanesulfonic acid (HEPES)–NaOH, 8.3 mM MgCl2, 1.7 mM ethylene glycol bis(2-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA), and 0.1% bovine serum albumin (BSA) (pH 7.4)) for 30 min at r.t. Final concentration of dimethyl sulfoxide (DMSO) in the reaction solution was 1%. Compounds were tested in duplicate in 96-well half-area plates (Corning) or 384-well OptiPlates (PerkinElmer, Inc.). We used an 8 concentration serial dilution dose response ranging from 100 µM to 10 pM compound concentrations. To start the reaction, 10 µL of substrate [3H] cGMP (final concentration 77 nM, PerkinElmer, Inc.) were added to a total volume of 40 µL. After 60 min at r.t., 20 µL of 20 mg/mL yttrium silicate SPA beads containing zinc sulfate were added to terminate the PDE reaction. After resting undisturbed for an additional 60 min, the assay plates were counted in a scintillation counter (PerkinElmer, Inc.) to allow calculation of the inhibition rate. Inhibition rate was calculated based on 0% control wells with enzyme and DMSO, and 100% control wells without enzyme. All IC50 values were obtained by fitting the results to the following 4 Parameter Logistic Equation:
where
A is the minimum
y value,
B is the maximum
y value,
C is Log(EC
50) value, and
D is the slope factor.
Human PDE Enzyme AssayPDE activities were measured using an SPA (GE Healthcare). To evaluate the inhibitory activity, 10 µL of serially diluted compounds were incubated with 20 µL of PDE enzymes (except for PDE1A) in assay buffer (50 mM HEPES–NaOH, 8.3 mM MgCl2, 1.7 mM EGTA, and 0.1% BSA (pH 7.4)) for 30 min at r.t. The PDE1A enzyme assay was performed in a different assay buffer (50 mM Tris–HCl, 8.3 mM MgCl2, 0.2 mM CaCl2, 0.1% BSA, and 30 nM Calmodulin (pH 7.5)). The final concentration of DMSO in the assay was 1%. Compounds were tested in duplicate in 96-well half-area plates (Corning). We used an 4 concentration serial dilution dose response ranging from 10 µM to 10 nM compound concentrations. To start the reaction, 10 µL of substrate ([3H] cGMP (final concentration 77 nM, PerkinElmer, Inc.) for PDE1A, 5A1, 6AB, 9A2, 10A2, and 11A4 or [3H] cAMP (final concentration 14.7 nM, PerkinElmer, Inc.) for PDE3A, 4D2, 7B, and 8A1) were added for a final assay volume of 40 µL. After 60 min incubation at r.t., 20 µL of 20 mg/mL yttrium silicate SPA beads containing ZnSO4 were added to terminate the PDE reaction. After resting undisturbed for more than 120 min, the assay plate was counted in a scintillation counter (PerkinElmer, Inc.) to allow calculation of the inhibition rate.
Transcellular Transport Study Using a Transporter-Expression SystemHuman MDR1-expressing LLC-PK1 cells were cultured with minor modifications to the method reported previously.57) The transcellular transport study was performed as reported previously.58) In brief, the cells were grown for 7 d in HTS Transwell 96-well permeable support (pore size 0.4 µm, 0.143 cm2 surface area) with polyethylene terephthalate membrane (Corning Life Sciences, Lowell, MA, U.S.A.) at a density of 1.125×105 cells/well. The cells were preincubated with M199 at 37°C for 30 min. Subsequently, transcellular transport was initiated by the addition of M199 either to apical compartments (75 µL) or to basolateral compartments (250 µL) containing 10 µM digoxin, 200 µM lucifer yellow (as a marker for the tightness of the monolayer), and 10 µM test compounds. The assay was terminated by the removal of each assay plate after 2 h. Aliquots (25 µL) from the opposite compartments were mixed with CH3CN containing alprenolol and diclofenac as internal standards, and then centrifuged. The compound concentrations in the supernatant were measured by LC-MS/MS. The apparent permeability (Papp) of test compounds in the receiver wells was determined and the efflux ratio (ER) for the MDR1 membrane permeability test was calculated using the following equation:
where
Papp, A to B is the apical-to-basal passive permeability-surface area product and
Papp, B to A is the basal-to-apical passive permeability-surface area product.
Phototoxicity TestPhototoxicity assay was carried out as described in the OECD guideline No. 43246 with some modifications for a high-throughput screening. BALB/c 3T3 cells were cultured at 37°C under 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum, 50 IU/mL penicillin and 50 mg/mL streptomycin. Cells were seeded at 2.5×103 cells/well in 384-well white plates, and cultured in DMEM supplemented with 10% fetal bovine serum, 2 mM L-glutamine, 1 mM sodium pyruvate, 50 IU/mL penicillin, and 50 µg/mL streptomycin for 1 d. Two 384-well plates per test compound (50 µM) in Earle’s Balanced Salt Solution (EBSS) supplemented with 1 mM HEPES were preincubated for 1 h. One of the two plates was irradiated (+UV) for 60 min with 1.4–1.7 mW/cm2 (5–6 J/cm2), whereas the other plate was kept in the dark (−UV). In both plates the treatment medium was replaced with the culture medium and after another 24 h of culture, the cell viability was determined by measuring the cellular ATP content. The cellular ATP content was measured using the Celltiter-Glo™ assay kit (Promega) following the manufacture’s instruction. ATP content was calculated as follows. ATP content (% of control)=(relative light unit (RLU) of test compound/RLU of 1% DMSO)×100.
Estimation of Log D at pH 7.4Log D7.4, which is the partition coefficient of the compounds between 1-octanol and aqueous buffer at pH 7.4, was measured using a chromatographic procedure based on a published method.59) The instruments utilized were a Waters Alliance 2795 HPLC system and a 2996 UV-Vis detector (Milford, MA, U.S.A.).
Thermodynamic Solubility Measurement Using the Shake-Flask MethodThe measurement of thermodynamic solubility was carried out as previously described.59) Briefly, the drug substances were weighed into Thomson filter vials (Chrom Tech, Inc., Minnesota, U.S.A.). JP1 (pH 1.2), JP2 (pH 6.8), and JP2-containing 20 mM GCDC were added to the vials. The vials were incubated at 37°C for 18 h and the resulting suspensions were filtered by compressing the vials. The drug concentration of the filtrates was determined using a UHPLC system.
Powder X-Ray Diffraction (PXRD) and Crystallinity CalculationPXRD patterns were collected using a RINT UltimaIV powder X-ray diffractometer (Rigaku Corp., Tokyo, Japan) according to previously described conditions.59) The peak intensities of the crystalline (Ic) and non-crystalline (Ia) fractions were integrated from the baseline collection according to Herman’s method.60,61) The crystallinity was calculated using the following equation with an autocrystallinity calculation software (Rigaku).61,62)
Protein Expression and PurificationThe PDE2A catalytic domain (578–919) was cloned into a pFastBac vector, for expression in Sf9 cells, utilizing an N-terminal 6×poly-histidine tag containing a TEV cleavage site. Large scale production of recombinant protein was carried out in Sf9 cells. The pellet from 10L of baculovirus infected Sf9 cells was resuspended in 600 mL lysis buffer containing 25 mM Tris pH 7.6, 1 M NaCl, 20 mM imidazole, 5% glycerol, and 3 Roche cOmplete Protease Inhibitor tablets. The cell suspension was homogenized with the Polytron PT-3100, centrifuged for 1 h at 13000 rpm (JA-14 rotor), and the clarified supernatant was brought to 800 mL with lysis buffer before batch binding with 10 mL of Probond Ni resin (Invitrogen) for 2 h at 4°C, rolling. The beads were collected by low speed centrifugation (3500 rpm with JS-4.2 rotor), loaded into a gravity column, and washed slowly overnight with 2 L of wash buffer containing 25 mM Tris pH 7.6, 1 M NaCl, 20 mM imidazole, 5% glycerol. The following day the protein was eluted with buffer containing 25 mM Tris pH 7.9, 50 mM NaCl, 250 mM imidazole, 10% glycerol. The 1.5 mL sample eluted from the Nickel capture step was brought to 9 mL with Mono Q buffer A containing 25 mM Tris pH 7.9, and 10% glycerol. After the full sample volume was bound to the Mono Q column, a salt gradient was applied from 0 M NaCl to ca. 800 mM NaCl in 40 mL. Fractions corresponding to the unphosphorylated protein (identified by MS with MW=40178 Da) were pooled for further purification by size-exclusion chromatography on a Superdex 200 column equilibrated in 1×TBS pH 7.4, 0.5 mM DTT, 1 mM EDTA, 10% glycerol. Peak SEC fractions were collected and concentrated to 12 mg/mL for crystallization.
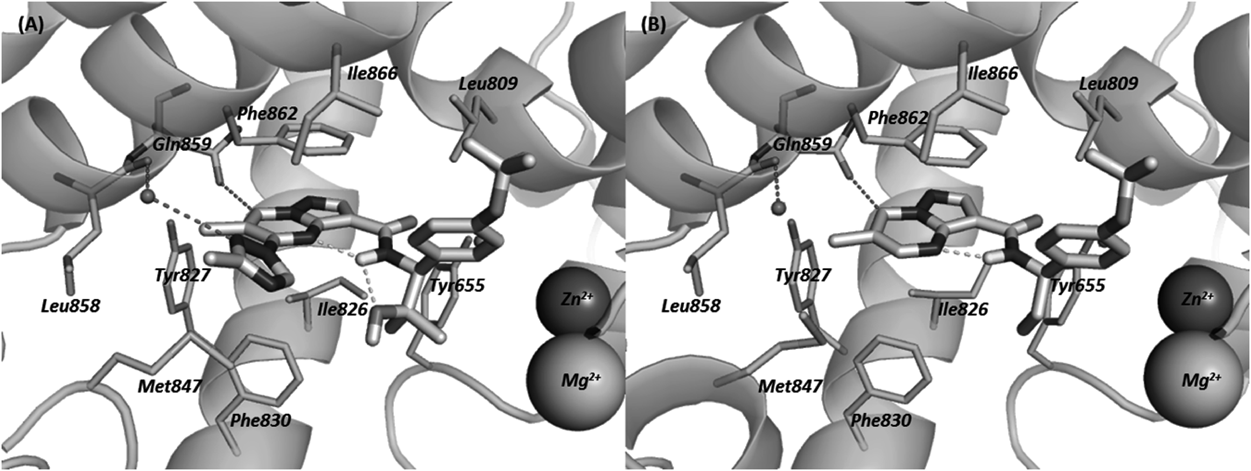
Crystallization and Structure DeterminationCrystals suitable for data collection were first grown using the vapor diffusion method in hanging drops at r.t. by adding 0.5 µL protein solution with 1 mM IBMX (1-methyl-3-(2-methylpropyl)-2,3,6,7-tetrahydro-1H-purine-2,6-dione) and 0.5 µL reservoir solution (30% PEG 3350, 0.1 M Tris pH 7.5, and 0.2 M MgCl2). PDE2A IBMX crystals were soaked in a drop containing 5 mM compound 20, 31% PEG 3350, 0.1 M Tris pH 7.5, and 0.2 M MgCl2 for 3 d. Crystals were transferred through a fresh cryo-protected soak drop immediately before being harvested and flash frozen in liquid nitrogen. X-Ray diffraction data was collected at ALS beamline 5.0.2 using a Pilatus3 6M (Dectris) detector from a single cryogenically protected crystal (100 K) at a wavelength of 1 Å. The crystals belong to the space group C121 and contain three enzyme molecules per asymmetric unit. X-Ray diffraction data was reduced using the HKL200063) software package. The structure was determined by molecular replacement with PHASER within the CCP4 program suite and refined with REFMAC.64) Several cycles of model building using MIfit65) and refinement using REFMAC were performed for improving the quality of the model. The coordinates and structure factors have been deposited in the Protein Data Bank with accession code 5VP1.
Animal ExperimentsThe care and use of animals and the experimental protocols were approved by the Experimental Animal Care and Use Committee of Takeda Pharmaceutical Company Limited.
Pharmacokinetic Analysis in Rat or Mouse Cassette DosingCompound 20 was administered intravenously (0.1 mg/kg) or orally (1 mg/kg) by cassette dosing to nonfasted male Crl : CD(SD)(IGS) rats (8 W, n=3) or male ICR mice (8 W, n=3). The combination for a cassette dosing was determined to avoid combinations of compounds with the same molecular weight. The solution of compounds in dimethylacetamide containing 50% (v/v) 1,3 butanediol at 0.1 mg/mL/kg was administered intravenously to isoflurane-anesthetized mice via femoral vein. The suspension of compounds in 0.5% methyl cellulose with water was used for vehicle (1 mg/kg) and was administered orally by gavage. After administration, blood samples were collected via tail vein by syringes with heparin at 5, 10, 15, 30 min, 1, 2, 4, and 8 h (intravenously (i.v.)) and 15, 30 min, 1, 2, 4, and 8 h (per os (p.o.)), and centrifuged to obtain the plasma fraction. The plasma samples were deproteinized by mixing with acetonitrile followed by centrifugation. The compound concentrations in the supernatant were measured by LC-MS/MS with a standard curve. Pharmacokinetic parameters were calculated by the non-compartmental analysis. The area under the concentration–time curve (AUC) and the area under the first moment curve (AUMC) were calculated using the linear trapezoidal method. The mean residence time (MRT) was calculated as AUMC/AUC. The total clearance (CLtotal) was calculated as doseiv/AUCi.v.. The volume of distribution (Vdss) was calculated as CLtotal×MRTi.v.. Oral bioavailability (F) was calculated as (AUCp.o./dosep.o.)/(AUCi.v./dosei.v.)×100.
Brain and Plasma Concentration in RatsCompound 20 was administered orally to Long–Evans rats (male, non-fasted, 7-week old) at 10 mg/kg. Blood and whole brain samples were collected 2 h after oral administration. The blood samples were centrifuged to obtain the plasma fraction. The brain samples were homogenized in saline to obtain the brain homogenate. Compound concentrations were measured in aliquots of rat plasma and brain, which were mixed well with acetonitrile containing an internal standard and then centrifuged. The supernatants were diluted with solvents for LC-MS/MS analysis (mobile phase A: 10 mM ammonium formate–formic acid (100 : 0.2, v/v), mobile phase B: acetonitrile–formic acid (100 : 0.2, v/v)). The diluted solutions were injected into an LC-MS/MS (API5000, AB Sciex, Foster City, CA, U.S.A.) equipped with a Shimadzu Shim-pack XR-ODS column (2.2 µm packing particle size, 2.0 mm ID×30 mm L) maintained at 50°C. The chromatographic separation was performed using gradient elution at a flow rate of 0.7 mL/min. The LC time program was as follows: Mobile phase B was held at 5% for 0.1 min, and increased linearly to 95% in 0.1 min. After maintaining B at 95% for another 0.8 min, it was decreased to 5% in 0.01 min, followed by re-equilibration for 0.59 min. The total cycle time for one injection was 1.6 min. Compound 20 was detected using multiple reaction monitoring mode and the transition m/z 490.01→240.96. Analyst™ software (version 1.4.2) was used for data acquisition and processing.
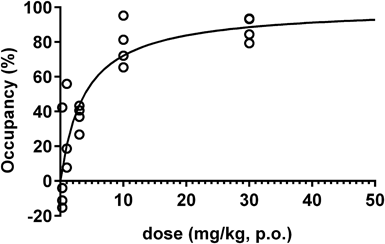
In Vivo Occupancy StudyIn vivo target occupancy study of compound 20 was conducted using LC-MS/MS. Compound 20 was suspended in 0.5% (w/v) methylcellulose in distilled water, and PF-0527043066) was dissolved in dimethylacetamide and 1,3-butanediol (1 : 1). Sprague-Dawley (SD) rats were pretreated with vehicle (p.o., n=6) or compound 20 (0.3, 1, 3, 10, 30 mg/kg, p.o., n=4 in each group) 2 h before sampling. PF-05270430 (0.1 mg/mL/kg) was administered by bolus intravenous injection via lateral tail vein 30 min before sampling. Rats were decapitated, and blood and brain samples (striatum as target tissue and cerebellum as reference tissue) were collected. Brain samples were weighed and saline was added (20% (w/v)), followed by homogenization with Lysing Matrix I beads (MP Biomedicals). Homogenized samples were stored at –30°C until quantification of tracer (PF-05270430) using LC-MS/MS. The supernatants were diluted with solvents for LC-MS/MS analysis (mobile phase A: 10 mM ammonium formate–formic acid (100 : 0.2, v/v), mobile phase B: acetonitrile–formic acid (100 : 0.2, v/v)). The diluted solutions were injected into an LC-MS/MS (API5000, AB Sciex) equipped with a Shimadzu Shim-pack XR-ODS (2.2 µm packing particle size, 2.0 mm ID×30 mm L) maintained at 50°C. The chromatographic separation was performed using gradient elution at a flow rate of 0.7 mL/min. The LC time program was as follows: Mobile phase B was held at 5% for 0.1 min, and increased linearly to 95% in 0.1 min. After maintaining B at 95% for another 0.8 min, it was decreased to 5% in 0.01 min, followed by re-equilibration for 0.59 min. The total cycle time for one injection was 1.6 min. PF-05270430 was detected using multiple reaction monitoring mode and the transition m/z 432→386. Analyst™ software (version 1.4.2) was used for data acquisition and processing. Specific tracer binding (BSP) in the striatum was represented as the difference between the tracer concentration in striatum and that in cerebellum. PDE2A occupancy was calculated using the following equation: Occupancy (%)=(BSP, base−BSP, drug)/BSP, base×100, where BSP, base and BSP, drug are the concentrations at baseline (vehicle treatment) and at drug treatment, respectively. Curve fitting of the saturation curve was carried out by nonlinear regression using GraphPad Prism 5.02 (GraphPad Software, Inc., San Diego, CA, U.S.A.).
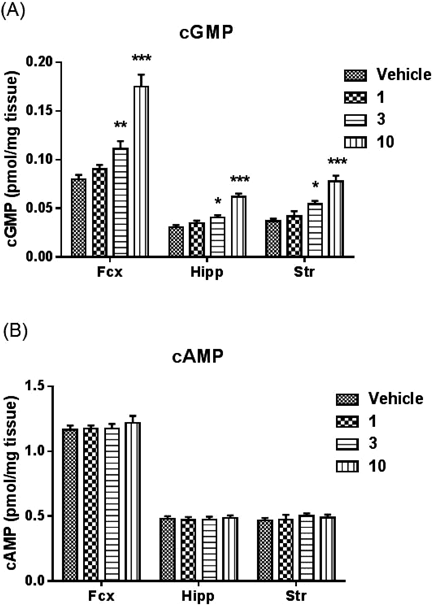
Measurement of Cyclic Nucleotide Contents in Rat BrainAnimalsFive-week-old male Long–Evans rats were purchased from Japan SLC, Inc. (Japan). The rats were housed in groups of 3/cage in a light-controlled room (12 h light/dark cycles with lights on at 07:00). Food and water were provided ad libitum. After a one week acclimation period, the six-week-old rats were used for experiments.
MeasurementsCompound 20 was suspended in 0.5% (w/v) methylcellulose in distilled water, and was administered in a volume of 2 mL/kg body weight for rats. Rats were administered orally with either vehicle or compound 20 (1, 3, 10 mg/kg) after >1 h of habituation. A microwave fixation system (Muromachi Kikai, Tokyo, Japan) was used to sacrifice unanesthetized rats by exposure of the head to the microwave beam at 2 h after administration of 20. Brain tissues were isolated and then homogenized in 0.5 mol/L HCl, followed by centrifugation. The concentration of cyclic nucleotides in the supernatant was measured using a cyclic AMP EIA kit or cyclic GMP EIA kit (Cayman Chemical, U.S.A.) following the manufacturer’s instructions. Values were expressed as pmol/mg tissue weight.
Step-through Passive Avoidance TaskThis task was performed as previously described67) with some modifications. This experiment was conducted in 7–8-week-old male Long–Evans rats. The apparatus (Brainscience idea, Osaka, Japan) consisted of an illuminated chamber (25×10×25 cm) connected to a dark chamber (30×30×30 cm) by a sliding door (8×8 cm). On the training day, each animal was subjected to a single pre-training trial 4–6 h before the acquisition trial. The rat was placed in the light chamber, and the sliding door was opened 30 s later. As soon as the rat entered the dark chamber with all four paws, the door was closed. The rat was then allowed to remain in the dark chamber for 30 s before being returned to its home cage. In the acquisition trial, the rat was placed in the light chamber, and the sliding door was opened. The time required for the rat to enter the dark chamber was then recorded. As soon as the rat entered the dark chamber, the door was closed, and an electric shock (0.5 mA, 3 s) was delivered from the floor grid. The rat was then returned to its home cage. The retention test was conducted 24 h later. The rat was again placed in the light chamber with the sliding door. After 30 s, the door was opened and the latency for the rat to cross over into the dark compartment was recorded. If the animal did not enter the dark chamber within 300 s, the retention test was terminated, and the animal was given a ceiling score of 300 s. Vehicle or compound 20 (3 mg/kg) was administered p.o. 2 h prior, and saline or MK-801 (0.1 mg/kg) was administered s.c. 30 min prior to the acquisition trial. The statistical significance of differences between group latency scores was determined by Wilcoxon’s test with significance set at p≤0.05.