Regular Articles
Synthesis of Tetrahydrobiphenylene via Pd(0)-Catalyzed C(sp2)–H Functionalization
2017 年 65 巻 12 号 p. 1167-1174
詳細
2017 年 65 巻 12 号 p. 1167-1174
Tetrahydrobiphenylene consists of cyclobutene fused with benzene and cyclohexene rings. In this paper, a direct method for synthesizing tetrahydrobiphenylenes based on a palladium (Pd)(0)-catalyzed C(sp2)–H functionalization was investigated. The developed method was applied to the synthesis of several tetrahydrobiphenylenes having an oxygen functionality at the ring juncture. The derivatization of a tetrahydrobiphenylene is also reported.
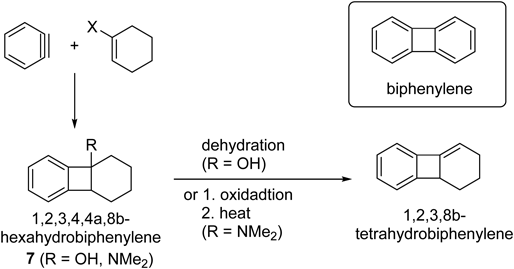
Biphenylenes, which have a structure consisting of a cyclobutadiene fused with two benzene rings (Chart 1), have been extensively studied to date.1) This class of compounds possesses an interesting aromaticity2) and the aryl–aryl C–C bond can be cleaved to give metallocyclic complexes.3–5) In contrast, the synthesis of partially hydrogenated biphenylenes, including tetrahydro- and hexahydrobiphenylenes has received little attention, despite these compounds showing interesting bioactivity. Caubère and colleagues have reported that di- and tetrahydrobiphenylenes showed cytotoxicity against human lymphoblasts and L1210 murine leukemia cell lines.6–8) For the synthesis of hexahydrobiphenylenes, a facial [2+2] cycloaddition of benzyne with an enamide9) and an enolate10) has been reported under strong base conditions (Chart 1). The resultant hexahydrobiphenylenes could be converted to tetrahydrobiphenylenes under dehydration conditions.10,11) Epoxidation of biphenylenes has also been reported for the synthesis of tetrahydro- and hexahydrobiphenylenes.12) However, these reported methods require harsh conditions and have only been applied to limited substrates. Additionally, there are no reports of direct methods to construct tetrahydrobiphenylenes. Thus, there is a need to improve the current synthetic methods for application to a wider range of substrates.
In recent decades, transition metal catalyzed C–H functionalization has been the focus of many groups as it can be used as a direct method for constructing molecules.13–20) Methods for the formation of benzocyclobutenes based on intramolecular C–H functionalization have been developed by the Catellani, Baudoin, Wolfe, and Martin groups.21–27) Recently, we have investigated Pd(0)-catalyzed benzylic C(sp3)–H functionalization for the synthesis of various heterocycles and tetrahydro-2H-fluorenes.28–41) In the synthesis of oxindoles, C(sp3)–H activation was favorable over competitive C(sp2)–H activation because of the presence of the sp2 nitrogen linkage29) (Chart 2a). In contrast, two methyl groups were required for the synthesis of tetrahydro-2H-fluorene through C(sp3)–H activation in the presence of the sp3 carbon linkage (Chart 2b).39) These results indicated that molecular flexibility is important for selectivity in this reaction. In this context, we attempted cyclization of a substrate having a C–H bond at the ortho position of the benzene ring, and observed the formation of tetrahydrobiphenylene through C(sp2)–H activation (Chart 2c). This reaction is potentially a direct synthetic method for the synthesis of tetrahydrobiphenylenes. The resultant tetrahydrobiphenylene can be derivatized into various hexahydrobiphenylenes because it contains several functional groups, including an olefin and allyl alcohol. In this report, we describe the development of a new synthetic method for tetrahydrobiphenylenes and derivatives.

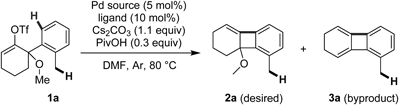
Initially, we investigated the reaction conditions for the formation of tetrahydrobiphenylene 2a from enol triflate 1a.42) Treatment of 1a with a catalytic amount of Pd(PPh3)4 (5 mol%), Cs2CO3 (1.1 equiv.), and PivOH (0.3 equiv.) in N,N-dimethylformamide (DMF) at 80°C gave tetrahydrobiphenylene 2a in 75% yield along with dihydrobiphenylene 3a (16%). The combination of Pd(OAc)2 (5 mol%) and PPh3 (10 mol%) resulted in a lower conversion rate and recovery of the starting material 1a (Table 1, entries 1 and 2). We next examined several ligands for suppressing the formation of the byproduct 3a. Use of an electron rich ligand, such as (2-furyl)3P, was not effective for suppression of the side reaction (entry 3). The reaction using bulky triaryl- and trialkyl-phosphine ligands, including (o-tolyl)3P, Ad2PnBu, and Cy3P·HBF4, hardly proceeded (entries 4–6), despite reports that bulky trialkyl-phosphine ligands were effective for C(sp3)–H functionalization.29) Using 5 mol% of 1,1′-bis(diphenylphosphino)ferrocene (dppf), a bidentate ligand, resulted in poor conversion (entry 7). In sharp contrast, the reaction with 10 mol% of dppf proceeded smoothly to give the desired 2a, suppressing the side reaction (entry 8). Dialkylbiarylphosphines, including dicyclohexyl(2′,4′,6′-triisopropyl-[1,1′-biphenyl]-2-yl)phosphine (XPhos) and dicyclohexyl(2′,6′-dimethoxy-[1,1′-biphenyl]-2-yl)phosphine (SPhos), were also effective for this transformation (entries 9 and 10). Because SPhos gave the best yield (93%), it was used for the further investigation of bases and additives. When other organic (Et3N) and inorganic (KOAc, Na2CO3, and K2CO3) bases were employed, the reaction hardly proceeded, or gave the desired product 2a in low yields (entries 11–14). The addition of PivOH was not essential, but it gave a more improved yield, than the use of 1-adamantanecarboxylic acid (AdCOOH) or cesium pivalate (CsOPiv) (entries 15–17). Unexpectedly, PivNHOH, which was effective for benzylic C(sp3)–H functionalization in the synthesis of oxindoles, completely suppressed the formation of the tetrahydrobiphenylene (entry 18). Therefore, the conditions using Pd(OAc)2 (5 mol%), SPhos or dppf (10 mol%), Cs2CO3 (1.1 equiv), and PivOH (0.3 equiv) in DMF at 80°C were determined to be the best conditions for the synthesis of tetrahydrobiphenylenes.
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Pd Source | Ligand | Base | Additive | Yielda) | ||
| 2a | 3a | 1a | |||||
| 1 | Pd(PPh3)4 | — | Cs2CO3 | PivOH | 75% | 16% | — |
| 2 | Pd(OAc)2 | PPh3 | Cs2CO3 | PivOH | 30% | 4% | 21% |
| 3 | Pd(OAc)2 | (2-furyl)3P | Cs2CO3 | PivOH | 50% | 13% | — |
| 4 | Pd(OAc)2 | (o-tolyl)3P | Cs2CO3 | PivOH | 3% | 3% | 80% |
| 5 | Pd(OAc)2 | Ad2PnBu | Cs2CO3 | PivOH | — | — | 85% |
| 6 | Pd(OAc)2 | Cy3P·HBF4 | Cs2CO3 | PivOH | — | — | 73% |
| 7 | Pd(OAc)2 | dppfb) | Cs2CO3 | PivOH | 8% | 1% | 77% |
| 8 | Pd(OAc)2 | dppf | Cs2CO3 | PivOH | 84% | 1% | — |
| 9 | Pd(OAc)2 | XPhos | Cs2CO3 | PivOH | 68% | 10% | — |
| 10 | Pd(OAc)2 | SPhos | Cs2CO3 | PivOH | 93% | 7% | — |
| 11 | Pd(OAc)2 | SPhos | Et3N | PivOH | 2% | — | 77% |
| 12 | Pd(OAc)2 | SPhos | KOAc | PivOH | 5% | 4% | 69% |
| 13 | Pd(OAc)2 | SPhos | Na2CO3 | PivOH | — | — | 81% |
| 14 | Pd(OAc)2 | SPhos | K2CO3 | PivOH | 40% | 8% | — |
| 15 | Pd(OAc)2 | SPhos | Cs2CO3 | None | 60% | 7% | — |
| 16 | Pd(OAc)2 | SPhos | Cs2CO3 | AdOOH | 69% | 10% | — |
| 17 | Pd(OAc)2 | SPhos | Cs2CO3 | CsOPiv | 77% | 19% | — |
| 18 | Pd(OAc)2 | SPhos | Cs2CO3 | PivNHOH | — | — | 89% |
a) Isolated yield. b) 5 mol% of dppf was used. Piv=pivaloyl, Ad=1-adamantyl, Cy=cyclohexyl, dppf=1,1′-bis(diphenylphosphino)ferrocene, XPhos=dicyclohexyl(2′,4′,6′-triisopropyl-[1,1′-biphenyl]-2-yl)phosphine, SPhos=dicyclohexyl(2′,6′-dimethoxy-[1,1′-biphenyl]-2-yl)phosphine.
The optimized conditions were applied to the synthesis of several tetrahydrobiphenylenes 2b–k. The substrate 1b42) having a methoxymethyl (MOM) group, instead of a methoxy group, was treated under the optimal conditions to give the cyclized product 2b in 84% yield (Table 2, entry 1). The reaction of substrates 1c and d, having bulkier ethyl and isopropyl substituents rather than the methyl group, gave the products 2c and d in 99 and 90% yields, respectively (entries 2 and 3). On the other hand, when substrate 1e having no substituent at the ortho position was employed, the yield of 2e was dramatically decreased (entry 4). In the case of 1f, having no MOM group, the cyclized product 2f was not obtained (entry 5). These results indicated that steric repulsion between the R1 and R3 substituents could be important. In the case of substrates 1g–k having substituents on the aromatic ring, the use of dppf, rather than SPhos, as a ligand was more effective. For example, while the reaction of 1g having an ester group gave a satisfactory result using SPhos, the yields for the reactions of substrates 1h–j decreased. In these cases, use of dppf improved the yields (entries 6–9). Additionally, tetrahydrobiphenylene 2k could be only accessed using dppf. Interestingly, no correlation could be found between the yield and the electronic state of the aromatic ring. In these investigations, tetrahydro-2H-fluorenes derived via C(sp3)–H activation were not observed.
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Substrate | R1 | R2 | R3 | Product | Yielda) | |
| SPhos | dppf | ||||||
| 1 | 1b | Me | H | OMOM | 2b | 84% | 87% |
| 2 | 1c | Et | H | OMOM | 2c | 99% | —c) |
| 3 | 1d | iPr | H | OMOM | 2d | 90% | —c) |
| 4 | 1e | H | H | OMOM | 2e | 8% | 22% |
| 5 | 1f | Me | H | H | 2f | 0%d) | —c) |
| 6 | 1g | Me | CO2Me | OMOM | 2g | 82% | —c) |
| 7 | 1h | Me | OMe | OMOM | 2h | 26% | 74% |
| 8 | 1i | Me | Me | OMOM | 2i | 65% | 77% |
| 9 | 1j | Me | CF3 | OMOM | 2j | 54%b) | 87% |
| 10 | 1k | Me | F | OMOM | 2k | 0% | 78% |
a) Isolated yield. b) The reaction time was 24 h. c) The reaction was not conducted. d) Starting material 1f was recovered (47%).
The proposed reaction mechanism is shown in Chart 3. The oxidative addition of enol triflate 1a to Pd(0) is followed by ligand exchange to give intermediate B through A. In comparison with our oxindole synthesis,29) the intermediate B should be more flexible because of the presence of the sp3 carbon at the ring juncture, and the Pd(II) center is close to an arene C(sp2)–H bond, rather than a methyl C(sp3)–H bond. Thus, C(sp2)–H activation proceeds, rather than C(sp3)–H activation, to give the palladacycle C. While electrophilic substitution to access intermediate C from A might be possible, it is unlikely to be the main pathway because no correlation between the yield and electronic state of the aromatic ring was observed. Finally, reductive elimination gives tetrahydrobiphenylene 2a. Byproduct 3a can be produced from the desired product through elimination of the methoxy group.

Because tetrahydrobiphenylene 2b having an oxygen functionality at the ring juncture can be accessed by only our method, we also investigated the possibility of functional group transformation of this compound (Chart 4). The compound 2b was unstable under acidic conditions; however, the MOM group was able to be removed by treatment with 80% aqueous acetic acid (AcOH) to give the allyl alcohol 4 in a moderate yield. Epoxidation of 2b with 3-chloroperoxybenzoic acid (mCPBA) gave compound 5 in 55% yield in a stereoselective manner. Similarly, hydroboration of 2b with thexylborane proceeded from only the β face to give the secondary alcohol 6 and tertiary alcohol 7 in 41 and 9% yields, respectively. The yields of these reactions were moderate presumably because of the instability of the starting material and products under the reaction conditions.

In summary, we have developed a new method for the synthesis of tetrahydrobiphenylenes based on a palladium (Pd)(0)-catalyzed C(sp2)–H functionalization. Applying this method, several tetrahydrobiphenylenes 2a–e, g–k having an oxygen functionality at the ring juncture could be accessed. We also investigated the reactivity of tetrahydrobiphenylene 2b. These results provide insights into a new aspect of the chemistry of biphenylenes.
Unless otherwise noted, all the reagents were purchased from chemical companies and used without further purification. All non-aqueous reactions were carried out under a positive pressure of argon in over-dried glassware. Analytical TLC was performed using Silica gel 60 plates (Merck, Darmstadt, Germany). Silica gel column chromatography was performed using Kanto silica gel 60 (particle size 63–210 µm, Kanto, Tokyo, Japan) and Chromatorex BW-300 (Fuji silysia, Aichi, Japan). Proton NMR (1H-NMR) spectra were recorded on a JNM-ECA 500 (JEOL, Tokyo, Japan) at 500 MHz or a JNM-AL 400 (JEOL) at 400 MHz. Chemical shifts were reported relative to Me4Si (δ 0.00) in CDCl3. Multiplicity was indicated by one or more of the following: s (singlet); d (doublet); t (triplet); q (quartet); m (multiplet); br (broad). Carbon NMR (13C-NMR) spectra were recorded on a JNM-ECA 500 at 126 MHz or a JNM-AL 400 at 101 MHz. Chemical shifts were reported relative to CDCl3 (δ 77.0). Infrared spectra were recorded on a FT/IR-4100 Fourier-transform IR spectrometer (JASCO, Tokyo, Japan) attenuated total reflectance (ATR). Low and high resolution mass spectra were recorded on JMS-700 mass spectrometer (JEOL) for FAB-MS and a LCMS-IT-TOF (Shimadzu, Kyoto, Japan) for electrospray ionization (ESI)-MS.
Preparation of Enol Triflate 1a2-Hydroxy-2-(o-tolyl)cyclohexan-1-one (8)To a solution of 2-[(trimethylsilyl)oxy]cyclohex-2-en-1-one43) (1.47 g, 7.97 mmol) in dry tetrahydrofuran (THF) (8.0 mL) was added o-tolylmagnesium bromide (8.90 mL, 0.9 M THF solution, 8.01 mmol, purchased from TCI) at room temperature under Ar. The reaction mixture was stirred at room temperature for 1.5 h, and then tetrabutylammonium fluoride (TBAF) (12.0 mL, 1.0 M in THF solution, 12.0 mmol, purchased from Aldrich) was added at the same temperature. The reaction mixture was stirred at room temperature for 2 h. The resultant mixture was diluted with Et2O and saturated aqueous NH4Cl. The aqueous phase was extracted with Et2O. The organic extracts were combined, washed with brine, and dried over Na2SO4. After filtration, the solution was concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (hexane/AcOEt=5/1) to give 2-hydroxy-2-(o-tolyl)cyclohexan-1-one (8) (1.53 g, 7.47 mmol, 94%) as a yellow oil: 1H-NMR (500 MHz, CDCl3) δ: 7.59 (1H, d, J=7.4 Hz), 7.29–7.23 (2H, m), 7.17 (1H, d, J=7.2 Hz), 4.48 (1H, s), 3.10 (1H, ddd, J=11.3, 6.1, 2.7 Hz), 2.51 (1H, m), 2.39 (1H, ddd, J=13.0, 13.0, 6.0 Hz), 2.14 (3H, s), 2.10 (1H, m), 1.87–1.74 (3H, m), 1.68 (1H, ddd, J=13.0, 13.0, 4.5 Hz); 13C-NMR (126 MHz, CDCl3) δ: 215.2, 138.0, 136.6, 132.6, 128.4, 127.0, 126.1, 81.5, 43.0, 39.0, 38.8, 30.3, 23.4, 20.7; IR (ATR) 3456, 2944, 2866, 1712, 1451, 1379, 1305, 1248, 1186, 1097, 1053, 1031 cm−1; HR-MS (ESI) m/z Calcd for C13H16O2Na [M+Na]+ 227.1043. Found 227.1051.
2-Methoxy-2-(o-tolyl)cyclohexan-1-one (9)To a solution of 2-hydroxy-2-(o-tolyl)cyclohexan-1-one (8) (391 mg, 1.91 mmol) in dry DMF (2 mL) was added a suspension of 60% sodium hydride (NaH) (111.2 mg, 2.78 mmol) in dry DMF (3 mL) slowly at 0°C. After stirring for 20 min at 0°C, the resultant mixture was treated with CH3I (288 µL, 4.60 mmol). The reaction mixture was stirred at room temperature for 17 h. The resultant mixture was diluted with Et2O and ice-water. The aqueous phase was extracted with Et2O. The organic extracts were combined, washed with brine, and dried over Na2SO4. After filtration, the solution was concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (hexane/AcOEt=20/1 to 10/1 to 5/1) to give 2-methoxy-2-(o-tolyl)cyclohexan-1-one (9) (262 mg, 1.20 mmol, 63%) as a colorless oil.: 1H-NMR (400 MHz, CDCl3) δ: 7.32 (1H, d, J=7.2 Hz), 7.24–7.18 (3H, m), 3.00 (3H, s), 2.85 (1H, m), 2.37–2.31 (3H, m), 2.20 (3H, s), 2.04–1.94 (2H, m), 1.87–1.77 (2H, m); 13C-NMR (101 MHz, CDCl3) δ: 210.0, 138.4, 135.6, 132.0, 128.1, 127.9, 125.4, 86.2, 50.0, 39.6, 36.4, 28.9, 21.6, 21.3; IR (ATR) 2940, 1722, 1485, 1308, 1252, 1207, 1150, 1132, 1099 cm−1; HR-MS (ESI) m/z Calcd for C14H18O2Na [M+Na]+ 241.1199. Found 241.1199.
1-Methoxy-2′-methyl-1,4,5,6-tetrahydro-[1,1′-biphenyl]-2-yl Trifluoromethanesulfonate (1a)To a solution of 2-methoxy-2-(o-tolyl)cyclohexan-1-one (9) (207.2 mg, 0.949 mmol) in dry THF (1.9 mL) was added lithium diisopropylamide (LDA (6.2 mL, 0.17 M THF solution, 1.05 mmol, freshly prepared from iPr2NH and nBuLi) at −78°C, and the resultant solution was stirred for 15 min. A solution of PhNTf2 (373.5 mg, 1.10 mmol) in dry THF (2.0 mL) was added, and the reaction mixture was stirred at 0°C for 1 h. The resultant mixture was diluted with AcOEt and water. The aqueous phase was extracted with AcOEt. The organic extracts were combined, washed with brine, and dried over Na2SO4. After filtration, the solution was concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (hexane) to give enol triflate 1a (206.5 mg, 0.589 mmol, 62%) as a yellow oil: 1H-NMR (400 MHz, CDCl3) δ: 7.40 (1H, m), 7.20–7.16 (3H, m), 6.23 (1H, t, J=4.0 Hz), 3.43 (3H, s), 2.47 (3H, s), 2.43–2.19 (3H, m), 2.12 (1H, m), 1.82 (1H, m), 1.55 (1H, m); 13C-NMR (101 MHz, CDCl3) δ: 148.3, 138.6, 135.8, 132.7, 127.7, 125.4, 121.7, 119.7, 116.5, 80.3, 52.1, 34.3, 24.5, 20.9, 18.5; IR (ATR) 2944, 1457, 1413, 1248, 1206, 1143, 1089, 1065, 1040 cm−1; HR-MS (FAB) m/z Calcd for C15H18F3O4S [M+H]+ 351.0878. Found 351.0873.
Preparation of Enol Triflate 1b2-(Methoxymethoxy)-2-(o-tolyl)cyclohexan-1-one (10)To a solution of 2-hydroxy-2-(o-tolyl)cyclohexan-1-one (8) (4.13 g, 20.2 mmol) in dry CH2Cl2 (100 mL) was added iPr2NEt (35.0 mL, 201 mmol) and MOMCl (15.4 mL, 203 mmol). The reaction mixture was refluxed for 9 h. After cooling to room temperature, the resultant mixture was poured into H2O and CHCl3. The aqueous phase was extracted with CHCl3. The organic extracts were combined, washed with brine, and dried over Na2SO4. After filtration, the solution was concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (hexane/AcOEt=5/1) to give 2-(methoxymethoxy)-2-(o-tolyl)cyclohexan-1-one (10) (4.25 g, 85%) as a pale yellow oil: 1H-NMR (400 MHz, CDCl3) δ: 7.47 (1H, m), 7.27–7.23 (2H, m), 7.20 (1H, m), 4.48 (2H, dd, J=13.8, 7.0 Hz), 3.38 (3H, s), 2.77–2.69 (2H, m), 2.34 (1H, m), 2.25 (1H, m), 2.19 (3H, s), 2.00–1.82 (4H, m); 13C-NMR (101 MHz, CDCl3) δ: 209.8, 138.4, 135.2, 132.3, 128.5, 128.0, 125.7, 92.3, 87.1, 56.2, 39.9, 39.5, 29.2, 22.5, 21.2; IR (ATR) 2940, 1724, 1487, 1449, 1308, 1252, 1232, 1208, 1163, 1145, 1132, 1101, 1055, 1013 cm−1; HR-MS (ESI) m/z Calcd for C15H20O3Na [M+Na]+ 271.1305. Found: 271.1305.
1-(Methoxymethoxy)-2′-methyl-1,4,5,6-tetrahydro-[1,1′-biphenyl]-2-yl Trifluoromethanesulfonate (1b)To a solution of 2-(methoxymethoxy)-2-(o-tolyl)cyclohexan-1-one (10) (56.1 mg, 0.226 mmol) in dry THF (0.50 mL) was added a solution of LDA (1.50 mL, 0.17 M THF solution, 0.255 mmol, freshly prepared from iPr2NH and nBuLi) at −78°C, and the resultant solution was stirred for 40 min. A solution of PhNTf2 (88.4 mg, 0.247 mmol) in dry THF (0.5 mL) was added, and the solution was stirred at 0°C for 5 h. The resultant solution was diluted with AcOEt and water. The aqueous phase was extracted with AcOEt. The organic extracts were combined, washed with brine, and dried over Na2SO4. After filtration, the solution was concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (hexane) to give enol triflate 1b (62.8 mg, 0.165 mmol, 73%) as a yellow oil: 1H-NMR (400 MHz, CDCl3) δ: 7.45 (1H, m), 7.20–7.16 (3H, m), 6.21 (1H, t, J=4.2 Hz), 5.07 (1H, d, J=7.6 Hz), 4.75 (1H, d, J=7.6 Hz), 3.51 (3H, s), 2.46 (3H, s), 2.42 (1H, m), 2.37–2.27 (2H, m), 2.15 (1H, m), 1.89 (1H, m), 1.61 (1H, m); 13C-NMR (101 MHz, CDCl3) δ: 148.0, 139.0, 135.2, 132.8, 127.7, 127.3, 125.6, 122.1, 119.8, 92.6, 80.2, 56.3, 36.4, 24.5, 21.2, 18.3; IR (ATR) 2947, 1484, 1457, 1416, 1249, 1212, 1147, 1045, 999, 932, 908 cm−1; HR-MS (ESI) m/z Calcd for C16H19F3OSNa [M+Na]+ 403.0798. Found 403.0792.
Synthesis of Tetrahydrobiphenylene 28b-Methoxy-8-methyl-1,2,3,8b-tetrahydrobiphenylene (2a)To a stirred solution of enol triflate 1a (28.1 mg, 0.0802 mmol), SPhos (3.3 mg, 0.0083 mmol), Cs2CO3 (28.7 mg, 0.0880 mmol), and PivOH (2.5 mg, 0.024.5 mmol) in DMF (0.40 mL) were added Pd(OAc)2 (0.9 mg, 0.0040 mmol). The reaction was stirred at 80°C for 4 h. After cooling to room temperature, the resulting mixture was filtered through a pad of celite using Et2O. The filtrate was washed with water and brine, and dried over Na2SO4. After filtration, the solution was concentrated under reduced pressure. The residue was purified by silica gel column chromatography (20% toluene/hexane) to give tetrahydrobiphenylene 2a (18.0 mg, 93%) containing dihydrobiphenylene 3a (1.4 mg, 7%) as a colorless oil: 1H-NMR (400 MHz, CDCl3) δ: 7.19 (t, 1H, J=6.0 Hz), 6.98 (d, 1H, J=6.0 Hz), 6.96 (d, 1H, J=6.4 Hz), 5.85 (dd, 1H, dd, J=3.8, 2.2 Hz), 3.17 (s, 3H), 2.39 (dd, 1H, J=18.4, 9.2 Hz), 2.28 (s, 3H), 2.24 (m, 1H), 2.17–2.09 (2H, m), 1.85 (m, 1H), 1.54 (m 1H); 13C-NMR (101 MHz, CDCl3) δ: 147.4, 147.4, 143.0, 134.0, 129.8, 128.7, 119.2, 115.7, 86.3, 52.6, 30.6, 29.7, 24.9, 18.9, 17.5; IR (ATR) 2933, 2896, 1474, 1455, 1435, 1349, 1330, 1229, 1158, 1119, 1072, 978 cm−1; HR-MS (ESI) m/z Calcd for C14H17O [M+H]+ 201.1274. Found 201.1268.
Methoxymethoxy-8-methyl-1,2,3,8b-tetrahydrobiphenylene (2b)Colorless oil, 253.4 mg, 87% yield (dppf); 1H-NMR (400 MHz, CDCl3) δ: 7.18 (1H, t, J=7.6 Hz), 6.98 (1H, d, J=7.6 Hz), 6.95 (1H, d, J=7.6 Hz), 5.83 (1H, dd, J=2.8, 4.8 Hz), 4.66 (1H, d, J=6.4 Hz), 4.62 (1H, d, J=6.8 Hz), 3.32 (3H, s), 2.45–2.38 (1H, m), 2.32–2.27 (4H, m), 2.22–2.11 (2H, m), 1.92–1.86 (1H, m), 1.58–1.50 (1H, m); 13C-NMR (101 MHz, CDCl3) δ: 147.5, 147.1, 143.2, 133.8, 129.9, 128.9, 118.5, 116.0, 92.9, 85.3, 55.4, 31.3, 24.8, 18.8, 17.4; IR (ATR) 2934, 1597, 1454, 1397, 1350, 1229, 1205, 1148, 1108, 1036, 963, 926, 882 cm−1; HR-MS (ESI) m/z Calcd for C15H19O2 [M+H]+ 231.1380. Found: 231.1382.
8-Ethyl-methoxymethoxy-1,2,3,8b-tetrahydrobiphenylene (2c)Colorless oil, 34.6 mg, 99% yield (SPhos); 1H-NMR (500 MHz, CDCl3) δ: 7.22 (1H, t, J=7.6 Hz), 7.00 (2H, dd, J=7.6, 4.7 Hz), 5.84–5.83 (1H, m), 4.61 (2H, dd, J=11.9, 6.7 Hz), 3.32 (3H, s), 2.66–2.61 (2H, m), 2.44–2.42 (1H, m), 2.30–2.27 (1H, m), 2.20–2.17 (2H, m), 1.90–1.89 (1H, m), 1.58–1.56 (1H, m), 1.24 (3H, t, J=7.6 Hz); 13C-NMR (126 MHz, CDCl3) δ: 147.7, 146.3, 143.4, 140.5, 130.1, 127.5, 118.4, 116.1, 92.7, 85.4, 55.5, 32.2, 25.4, 24.8, 18.8, 14.7; IR (ATR) 2937, 1748, 1472, 1107, 1039 cm−1; HR-MS (ESI) m/z calcd for C16H20O2Na: [M+Na]+ 267.1361. Found 267.1356.
8-Isopropyl-methoxymethoxy-1,2,3,8b-tetrahydrobiphenylene (2d)Pale yellow oil, 31.4 mg, 90% yield (SPhos); 1H-NMR (500 MHz, CDCl3) δ: 7.25–7.22 (1H, m), 7.03–7.01 (2H, m), 5.84–5.83 (1H, m), 4.62 (1H, d, J=6.6 Hz), 4.54 (1H, d, J=6.6 Hz), 3.33 (3H, s), 2.96–2.90 (1H, m), 2.44–2.43 (1H, m), 2.30–2.28 (1H, m), 2.20–2.18 (2H, m), 1.91–1.90 (1H, m), 1.61–1.55 (1H, m), 1.26 (6H, dd, J=8.0, 7.2 Hz); 13C-NMR (126 MHz, CDCl3) δ: 147.8, 145.6, 145.2, 143.5, 130.1, 125.9, 118.2, 116.0, 92.3, 85.4, 55.5, 32.9, 31.6, 24.7, 23.5, 23.1, 18.9; IR (ATR) 2957, 1147, 1105, 1031 cm−1; HR-MS (ESI) m/z Calcd for C17H22O2Na: [M+Na]+ 281.1517. Found 281.1512.
Methyl-methoxymethoxy-4-methyl-4b,5,6,7-tetrahydrobiphenylene-2-carboxylate (2g)Yellow oil, 31.1 mg, 82% yield (SPhos); 1H-NMR (500 MHz, CDCl3) δ: 7.72 (1H, s), 7.64 (1H, s), 5.94–5.93 (1H, m), 4.66 (1H, d, J=6.9 Hz), 4.61 (1H, d, J=6.9 Hz), 3.90 (3H, s), 3.29 (3H, s), 2.46–2.41 (1H, m), 2.34 (3H, s), 2.31–2.28 (1H, m), 2.22–2.14 (2H, m), 1.92–1.91 (1H, m), 1.54–1.51 (1H, m); 13C-NMR (126 MHz, CDCl3) δ: 167.3, 152.4, 147.5, 142.1, 133.9, 131.7, 130.7, 120.4, 117.1, 93.0, 84.9, 55.5, 52.1, 31.1, 24.8, 18.6, 17.3; IR (ATR) 2949, 1722, 1415, 1207, 1042 cm−1; HR-MS (ESI) m/z Calcd for C17H20O4Na: [M+Na]+ 311.1259. Found 311.1254.
6-Methoxy-methoxymethoxy-8-methyl-1,2,3,8b-tetrahydrobiphenylene (2h)Colorless oil, 33.0 mg, 74% yield (dppf); 1H-NMR (500 MHz, CDCl3) δ: 7.84 (2H, d, J=8.0 Hz), 7.58 (1H, d, J=8.0 Hz), 6.25–6.24 (1H, m), 5.09 (1H, d, J=7.7 Hz), 4.76 (1H, d, J=8.0 Hz), 3.91 (3H, s), 3.52 (3H, s), 2.49–2.45 (4H, m), 2.37–2.28 (2H, m), 2.12–2.07 (1H, m), 1.91–1.89 (1H, m), 1.63–1.61 (1H, m); 13C-NMR (126 MHz, CDCl3) δ: 161.5, 148.3, 142.7, 139.1, 135.3, 118.0, 115.7, 101.0, 92.7, 84.4, 55.37, 55.33, 31.5, 24. 7, 18.8, 17.5; IR (ATR) 2936, 1596, 1475, 1138, 1040 cm−1; HR-MS (ESI) m/z Calcd for C16H20O3Na: [M+Na]+ 283.1310. Found 283.1305.
Methoxymethoxy-6,8-dimethyl-1,2,3,8b-tetrahydrobiphenylene (2i)Colorless oil, 20.6 mg, 77% yield (dppf); 1H-NMR (500 MHz, CDCl3) δ: 6.83 (1H, s), 6.79 (1H, s), 5.79–5.78 (1H, m), 4.66 (1H, d, J=6.9 Hz), 4.62 (1H, d, J=6.9 Hz), 3.33 (3H, s), 2.42–2.40 (1H, m), 2.31 (3H, s), 2.26–2.24 (4H, m), 2.20–2.12 (2H, m), 1.89–1.86 (1H, m), 1.53–1.50 (1H, m); 13C-NMR (126 MHz, CDCl3) δ: 147.6, 144.1, 143.0, 139.9, 133.5, 129.8, 117.9, 116.6, 92.8, 84.8, 55.4, 31.4, 24. 7, 21.9, 18.7, 17.3; IR (ATR) 2933, 1591, 1453, 1151, 1029 cm−1; HR-MS (ESI) m/z Calcd for C16H20O2Na: [M+Na]+ 267.1361. Found 267.1356.
8b-(Methoxymethoxy)-8-methyl-6-(trifluoromethyl)-1,2,3,8b-tetrahydrobiphenylene (2j)Yellow oil, 34.8 mg, 87% yield (dppf); 1H-NMR (500 MHz, CDCl3) δ: 7.23 (2H, s), 5.96 (1H, dd, J=4.5, 2.4 Hz), 4.66 (1H, d, J=6.9 Hz), 4.61 (1H, d, J=6.9 Hz), 3.31 (3H, s), 2.45 (1H, m), 2.35 (3H, s), 2.30 (1H, ddd, J=12.6, 4.2, 2.4 Hz), 2.24–2.14 (2H, m), 1.92 (1H, m), 1.51 (1H, ddd, J=13.0, 13.0, 4.2 Hz); 13C-NMR (126 MHz, CDCl3) δ: 150.8, 147.6, 142.0, 134.6, 132.1 (q, J=31.2 Hz), 125.8 (q, J=3.5 Hz), 124.2 (q, J=272.8 Hz), 121.1, 113.0 (q, J=3.5 Hz), 92.9, 84.9, 55.5, 31.1, 24.9, 18.6, 17.4; IR (ATR) 2948, 2889, 1327, 1307, 1201, 1160, 1127, 1042 cm−1; HR-MS (FAB) m/z Calcd for C16H17F3O2Na: [M+Na]+ 321.1078. Found 321.1077.
6-Fluoro-methoxymethoxy-8-methyl-1,2,3,8b-tetrahydrobiphenylene (2k)Pale yellow oil, 36.3 mg, 78% yield (dppf); 1H-NMR (400 MHz, CDCl3) δ: 6.70–6.67 (2H, m), 5.87–5.86 (1H, m), 4.65 (1H, d, J=6.9 Hz), 4.61 (1H, d, J=6.9 Hz), 3.31 (3H, s), 2.47–2.40 (1H, m), 2.28–2.27 (4H, m), 2.21–2.14 (2H, m), 1.92–1.88 (1H, m), 1.53–1.47 (1H, m); 13C-NMR (101 MHz, CDCl3) δ: 165.2, 163.2, 148.4 (d, J=6.7 Hz), 142.3 (d, J=35.2 Hz), 136.3 (d, J=6.7 Hz), 119.7, 115.9 (d, J=19.1 Hz), 103.7 (d, J=18.2 Hz), 92.7, 84.2, 56.4, 31.4, 24.8, 18.8, 17.4; IR (ATR) 2939, 1586, 1467, 1348, 1107, 1030 cm−1; HR-MS (ESI) m/z Calcd for C15H17FO2Na: [M+Na]+ 271.1110. Found 271.1105.
Removal of MOM Group of Compound 2bCompound 2b (30.3 mg, 0.132 mmol) was dissolved in 80% aq. AcOH (1.3 mL). The solution was stirred at room temperature for 10 h, and 50°C for 16 h. After cooling to room temperature, the resulting solution was diluted with EtOAc, washed with satd. aq. NaHCO3, and dried over Na2SO4. After filtration and concentration under reduced pressure, the residue was purified by silica gel column chromatography (10–20% EtOAc/hexane) to give compound 4 (9.0 mg, 37%) as a colorless oil: 1H-NMR (400 MHz, CDCl3) δ: 7.18 (1H, t, J=7.5 Hz), 7.00 (1H, d, J=7.5 Hz), 6.97 (1H, d, J=7.5 Hz), 5.78 (1H, dd, J=4.6, 2.3 Hz), 2.42 (1H, m), 2.31 (3H, s), 2.29–2.10 (4H, m), 1.90 (1H, m), 1.62 (1H, ddd, J=12.6, 12.6, 4.3 Hz); 13C-NMR (101 MHz, CDCl3) δ: 150.5, 147.1, 145.0, 132.8, 129.8, 129.2, 117.7, 116.7, 80.9, 31.0, 25.0, 18.5, 17.1; IR (ATR) 3380, 3042, 2938, 2866, 1596, 1453, 1375, 1102, 1036, 984 cm−1; HR-MS (ESI) m/z Calcd for C13H15O: [M+H]+ 187.1117. Found 187.1115.
Epoxydation of Compound 2bTo a solution of compound 2b (84.0 mg, 0.365 mmol) in CH2Cl2 (5 mL) was added NaHCO3 (66.2 mg, 0.788 mmol) and mCPBA (77% purity, 90.5 mg, 0.403 mmol) at 0°C. The resultant mixture was stirred at 0°C overnight. The resulting mixture was diluted with EtOAc, and washed with satd. aq. NaHCO3 and brine. The organic layer was dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (5% EtOAc/hexane) to give compound 5 (49.6 mg, 55%) as a colorless oil: 1H-NMR (400 MHz, CDCl3) δ: 7.28 (1H, t, J=7.8 Hz), 7.15 (1H, d, J=7.8 Hz), 7.01 (1H, d, J=7.8 Hz), 4.82 (1H, d, J=6.7 Hz), 4.71 (1H, d, J=6.7 Hz), 3.55 (1H, d, J=2.3 Hz), 3.37 (3H, s), 2.34 (3H, s), 2.30–2.18 (2H, m), 1.80–1.68 (2H, m), 1.56–1.43 (2H, m); 13C-NMR (101 MHz, CDCl3) δ: 146.2, 145.0, 133.8, 130.8, 130.4, 119.0, 93.2, 86.7, 67.4, 58.4, 55.8, 32.2, 25.8, 17.6, 16.3; IR (ATR) 2938, 1610, 1465, 1151, 1116, 1034 cm−1; HR-MS (ESI) m/z Calcd for C15H18O3Na: [M+Na]+ 269.1148. Found 269.1134.
Hydroboration of Compound 2bTo a solution of compound 2b (30.0 mg) in dry THF (1 mL) was added thexylborane solution (0.5 M in THF, 0.39 mL, 0.195 mmol, prepared from BH3·SMe2 (2.0 M in THF, 1.25 mL) and 2,3-dimethyl-2-butene (0.30 mL)) at 0°C. The resultant mixture was stirred at 0°C for 110 min. The solution was treated with 3 M aq. NaOH (0.39 mL) followed by 30% aq. H2O2, and stirred at room temperature for 2.5 h. The resulting mixture was diluted with EtOAc, and washed with satd. aq. NH4Cl and brine. The organic layer was dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (25–40% EtOAc/hexane) to give compound 7 (2.9 mg, 9%) and then compound 6 (13.8 mg, 41%) as a colorless oil: compound 6: 1H-NMR (400 MHz, CDCl3) δ: 7.19 (1H, t, J=7.5 Hz), 7.03 (1H, d, J=7.5 Hz), 6.96 (1H, d, J=7.5 Hz), 4.84 (1H, d, J=7.2 Hz), 4.75 (1H, d, J=7.2 Hz), 4.26 (1H, ddd, J=4.6, 4.6, 4.6 Hz), 3.68 (1H, d, J=4.6 Hz), 3.44 (3H, s), 2.34 (1H, m), 2.25 (3H, s), 1.93 (1H, m), 1.77 (1H, m), 1.63–1.58 (2H, m), 1.25 (1H, m); 13C-NMR (101 MHz, CDCl3) δ: 144.9, 142.7, 133.3, 129.5, 129.0, 119.6, 92.7, 83.2, 69.7, 56.4, 55.5, 30.4, 27.8, 17.1, 16.8; IR (ATR) 3423, 2936, 1729, 1612, 1450, 1349, 1146, 1031 cm−1; HR-MS (ESI) m/z Calcd for C15H20O3Na: [M+Na]+ 271.1305. Found 271.1280; compound 7: 1H-NMR (400 MHz, CDCl3) δ: 7.28 (1H, t, J=7.5 Hz), 7.12 (1H, d, J=7.5 Hz), 7.10 (1H, d, J=7.5 Hz), 4.84 (1H, d, J=6.4 Hz), 4.71 (1H, d, J=6.4 Hz), 4.41 (1H, s), 3.33 (3H, s), 2.28 (3H, s), 2.12–2.09 (2H, m), 2.05–1.93 (2H, m), 1.66–1.49 (3H, m), 1.10 (1H, m), 0.96 (1H, m); 13C-NMR (101 MHz, CDCl3) δ: 150.5, 141.3, 135.1, 130.4, 130.1, 119.7, 93.4, 90.8, 80.6, 55.6, 30.9, 29.1, 17.7, 17.2, 16.7; IR (ATR) 3435, 2940, 1728, 1608, 1467, 1344, 1148, 1040 cm−1; HR-MS (ESI) m/z Calcd for C15H20O3Na: [M+Na]+ 271.1305. Found 271.1286.
We thank Mrs. T. Kimura, T. Sakaguchi, Y. Tokuhiro, and Ms. N. Kato for checking reproducibility and spectra data. This work was supported by the Japan Society for the Promotion of Science (JSPS) KAKENHI (Grant No. JP17H05051), the Platform Project for Supporting Drug Discovery and Life Science Research from Japan Agency for Medical Research and Development (AMED). We thank Victoria Muir, Ph.D., from Edanz Group (www.edanzediting.com/ac) for editing a draft of this manuscript.
The authors declare no conflict of interest.
The online version of this article contains supplementary materials.
