Abstract
In this study, we developed highly dispersible polylactic glycolic acid (PLGA) copolymer microparticles (MRPs) in aqueous fluid. A solution containing both dissolved aripiprazole as a model drug and PLGA were spray-dried to make MRPs. The resultant MRPs were further co-processed with water-soluble additives and a surfactant to improve their dispersion behavior. The granules containing MRPs and additives, termed granulated microparticles (G-MRPs) were prepared by a newly established drop freeze-drying technique. The physicochemical properties of MRPs and G-MRPs were evaluated as a long-acting release depot injectable. The MRPs were spherical particles with diameters of approximately 1 to 20 µm and strongly assembled to one another in the aqueous phase, forming large aggregations. In contrast, the G-MRPs were spherical granules with diameters of approximately 200 to 400 µm that displayed a microparticles-in-granule structure in which small MRPs were embedded in the porous matrix inside the granules. When the G-MRPs were placed in water, the porous matrix base was immediately dissolved, and each embedded MRP was individually released, thus inducing monodispersion and significantly improved dispersibility. The excellent dispersibility was attributed to the water-soluble porous network structure mainly composed of D-mannitol and the steric hindrance effects derived from the polymeric molecular chains. These properties may give rise to the excellent passage of PLGA microparticles through needles for use in depot formulation suspensions. A crystalline evaluation of the G-MRPs suggested that the drug and PLGA molecularly interacted and that their thermodynamic stability was improved.
Injectable products composed of biocompatible and biodegradable polylactic glycolic acid (PLGA) copolymer microparticles have been available on the market for long term-acting therapy. Several manufacturing methods are well known for commercially producing PLGA microparticles products. Chen et al. reported that Sandostatin LAR® Depot of somatostatin were prepared by the oil/water (O/W) single emulsion method.1) Besides, Nutropin Depot® of human Growth Hormone (hGH) was developed by the W/O/W phase separation method mentioned by Jones et al.2) Further, a one-month sustained-release depot formulation of leuprorelin acetate (Leuplin®) of PLGA was developed by the W/O/W emulsion solvent-evaporation method.3–6) Since it was marketed in the U.S.A. in 1989, Leuplin® has been used worldwide, and a recently developed six-month formulation that lasts up to 24 weeks currently contributes to improving the QOL of many patients.7,8) However, such formulations are manufactured through multiple processes, such as by dissolution of the active pharmaceutical ingredient and additives, filtration, primary and secondary emulsification, evaporation of the organic solvent, centrifugation, lyophilization, pulverization, and powder filling. Enormous effort and expense are required for quality control, as all processes must be completed in a sterile environment.9) In addition to this complexity, a disadvantage of these processes is that the particle size distribution (PSD) cannot be easily controlled because the emulsion manufacturing method utilizes high shear mixing.10)
Recently, the spray-drying method has attracted increased attention for use in drug manufacturing.11) Spherical microparticles consisting of a drug and a controlled-release polymer can be prepared in one step via a spray-drying process, and the resultant particles have a narrow PSD compared with those fabricated by the emulsion manufacturing method. Thus, the spray-drying process is advantageous for easily controlling particle size.12) However, few injectable formulations manufactured by the spray-drying method are available on the market. Regarding PLGA formulations, Parlodel manufactured by Sandoz is the only injectable formulation on the market, and its use remains low.13) Its limited use may be because spray-drying is associated with risks that some of the organic PLGA solvent (e.g., dichloromethane) is left behind as residual solvent or that the PLGA particles may become electrostatic and easily adhere to neighboring structures, thus contributing to poor handling. PLGA formulations that have been manufactured via the emulsion manufacturing method, such as Leuplin®, have been widely sold; however, for use in medical fields, these formulations are characterized by an unresolved issue that arises at the time of administration. To prepare the suspension at the time of administration, the PLGA formulation is dispersed in an injection solution. However, PLGA particles of a single PLGA formulation exhibit poor dispersibility in the aqueous phase, which poses a risk that particles could become aggregated and clog the needle upon administration, as it is difficult to maintain a uniform dispersion of the suspension after the PLGA particles are mixed in an aqueous medium. Thus, to prevent aggregation, a vehicle with a dedicated formulation containing surface active agents and high viscosity additives is provided as an attached kit with the marketed formulations. Although these suspensions are expected to highly improve the dispersibility and passage of the formulations through the needle, very few reports of the delivery efficacy are available.14)
Based on this background, we adopted the spray-drying method to produce PLGA microparticles (MRPs). Furthermore, to improve the dispersibility in the MRP injection solutions, we subjected the spray-dried MRPs to a secondary process, i.e., the freeze-drying method. Thus, instead of obtaining a typical cake-like powder in the vial, we sought to obtain a granulate with favorable handling properties like a powder. Specifically, we sought to process the PLGA microparticles to obtain superior granular powder properties, yielding a powder that is suitable for injections and that can be pre-filled into syringes and marketed as a ready-to-use formulation. For the manufacturing process, the MRPs obtained via the spray-drying method were suspended in an aqueous solution of a water-soluble additive and a surfactant. Then, the suspension was rapidly dripped into liquid nitrogen in the form of liquid drops. The resultant frozen particles were freeze-dried to prepare granules containing MRP (G-MRPs). We further investigated the formulation to develop MRPs that have the potential for spontaneous dispersion without aggregation when mixed with the injection solution.
Aripiprazole (ARP) was selected15) as a model drug, as ARP is used to treat schizophrenia patients and should have a sustained release upon administration. ARP is used as a maintenance therapy during the treatment of schizophrenia, and it is important that its formulation can be immediately dispersed and administered to schizophrenic patients. Thus, we considered ARP as an ideal model drug for developing a ready-to-use formulation with excellent dispersibility in an injection vehicle and re-dispersibility.
Experimental
MaterialsARP as an active pharmaceutical drug was a gift from Isekyu Co., Ltd. (Nagoya, Japan). Poly(DL-lactide-co-glycolide) (PLGA 75–50, lactide : glycolide=75 : 25, molecular weight (MW) 50000) was provided by Mitsui Chemical Co., Ltd. (Tokyo, Japan). Poly(DL-lactide-co-glycolide) is briefly described as PLGA in this report. D-Mannitol (MNT; Mannit P) was provided by Mitsubishi Shoji Foodtech Co., Ltd. (Tokyo, Japan). Hydroxypropyl cellulose (HPC; HPC-SL), polyvinyl alcohol (PVA; EG-40P) and sodium lauryl sulfate (SLS) were provided by Nippon Soda Co., Ltd. (Tokyo, Japan), Nippon Gousei Chemical Co., Ltd. (Tokyo, Japan) and Wako Pure Chemical Industries, Ltd. (Osaka, Japan), respectively. Water-soluble additives, such as MNT, HPC, and PVA, are referred to as “carrier” in this paper. All other chemicals and solvents were of analytical reagent grade, and deionized distilled water was used throughout the study.
Manufacturing InstrumentsA spray dryer (B-290; Nihon Büchi Co., Ltd., Tokyo, Japan) was used to prepare the MRPs in this study. An encapsulator (B-390; Nihon Büchi Co., Ltd.) was used to form droplets composed of the MRP suspension. A freeze dryer (FD-550; Tokyo Rikakikai Co., Ltd., Tokyo, Japan) was used to lyophilize the resultant frozen droplets.
Spray-Drying Process for MRP PreparationThe spray-drying process using the B-290 spray dryer is illustrated in Fig. 1. Dichloromethane was used as a spray solvent to dissolve both ARP and PLGA. The formulation of the spray solutions in this study is described in Table 1. The ratio of drug to PLGA was set according to a previous study in which sustained-release microparticles were developed.16) The dichloromethane solution was sprayed through a 2-fluid nozzle (0.5 mmφ) to prepare the MRPs containing ARP. The operating parameters were set to the standard conditions considering the solvent with the lower boiling point, as listed in Table 2.
Table 1 Formulation of the Spray Solution of MRPs with ARP Supplied to the Spray-Drying Process
| Material | Weight (g) |
|---|
| ARP | 40 |
| PLGA | 15 |
| Dichloromethane | q.s. |
| Total | 1000 |
Table 2 Operating Conditions of the Spray-Drying Process
| Parameter | Condition |
|---|
| Inlet air temperature | 51°C |
| Outlet air temperature | 36°C |
| Spray rate | 20 mL/min |
| Nozzle cooling | On |
| Nozzle aperture | 1.4 mmϕ |
| Spray flow | 300 L/h |
| Spray gas | Nitrogen gas |
| Aspirator setting for gas flow | 35 m3/h |
To prevent aggregation of the hydrophobic MRPs in water, water-soluble HPC, PVA, and D-mannitol were selected as additives (hereafter referred to as the carrier). SLS was also blended as a surfactant to improve the water wettability. Five blended formulations with different additives and components of the carrier were examined (Table 3). In the carrier, HPC and PVA were loaded as a dispersion-improving agent for MRP, whereas D-mannitol was loaded as wetting-improving agent for the granules. Among the formulations, only G-MRP 50%MNT formulation contained additives with all three functions (a dispersing agent, a wetting agent, and a surfactant). The 5 G-MRP symbols correspond to the weight ratio of the additives to the total solid components in the G-MRPs. For example, the G-MRP 50%HPC corresponds to 50% (w/w) because the carrier includes 570 mg HPC and 30 mg SLS in the total solid component of 1200 mg. Each additive was dissolved, and MRPs were suspended in purified water as the dropping liquid to prepare the suspension with 5% (w/v) of solid concentration. While vibrating, the MRP suspension was dripped into liquid nitrogen using the liquid droplet forming function of the encapsulator device while stirring with a magnetic stirrer. A schematic diagram of the manufacturing process is shown in Fig. 1. To obtain granules of 450 to 600 µm in size, the suspension was fed at a high speed rate of 5 mL/min using a nozzle with an inner diameter of 300 µm at a 700 Hz dropwise vibration frequency and 1200 V electrode voltage. The ice particles that were quickly frozen after dropping were lyophilized (FD-550, Tokyo Rikakikai Co., Ltd.) to obtain a composite particle composed of the MRPs and the carrier. Lyophilization was performed for 24 h at −30°C for all formulations, and then the formulations were gradually heated to 30°C below 8 Pa. The particles were termed as granulated microparticles because the particles formed granules. Hereafter, the abbreviation G-MRPs refers to the granules that include MRPs. The collected G-MRPs were stored in a refrigerator in glass vials until the physical chemical evaluation.
Table 3 G-MRP Formulations of the Granulated Microparticles
| Symbol | | G-MRP 50%HPC | G-MRP 20%HPC | G-MRP 50%PVA | G-MRP 20%PVA | G-MRP 50%MNT |
|---|
| MRP | (mg) | 600 | 600 | 600 | 600 | 600 |
| HPC | (mg) | 570 | 142.5 | — | — | — |
| PVA | (mg) | — | — | 570 | 142.5 | 71.25 |
| SLS | (mg) | 30 | 7.5 | 30 | 7.5 | 3.75 |
| D-Mannitol | (mg) | — | — | — | — | 525 |
| Purified water | (mL) | 24 | 15 | 24 | 15 | 24 |
| Total solid component amount | (mg) | 1200 | 750 | 1200 | 750 | 1200 |
| Solution | (mL) | 24 | 15 | 24 | 15 | 24 |
| Solid component concentration | (%) | 5.0 | 5.0 | 5.0 | 5.0 | 5.0 |
The MPRs and G-MRPs were coated using platinum sputtering equipment (JFC-1600; JEOL Ltd., Tokyo, Japan), and the particle morphology and surface conditions were observed using a scanning electron microscopy (SEM JSM-6060; JEOL Ltd.). The PSDs of the MRPs and G-MRPs were measured via a laser diffraction scattering method using a diffractometer (LMS-30; Seishin Enterprise Co., Ltd., Tokyo, Japan). Measurements were performed by dispersing the particles in compressed air (0.4 MPa), and then the PSDs of the MRPs and G-MRPs (10% particle size; D10, 50% particle diameter; D50, 90% particle diameter; D90) were measured via a dry method. In addition, the images obtained from the SEM photographs were analyzed using an electronic image analyzer (Luzex AP, Nireco, Tokyo, Japan), and the Heywood diameters of 500 particles were measured based on the cumulative PSD diagram. The cumulative 50% particle size (median diameter) was read out from the cumulative distribution curve based on the volume rule and determined as the geometric average diameter (geoD50).
Dispersibility of the MRPs and G-MRPs in the Aqueous PhaseThe PSD dispersed in the aqueous medium was measured using a laser diffractometer (LMS-30, Seishin Enterprise). The MRPs or G-MRPs powder was directly placed into a dispersing medium under weak agitation with a micro-stirrer. Water for injection for use as the dispersion medium. The PSD of the dispersed MRPs were measured at each preset time (0.5, 1, 1.5, 2, 2.5, 3, 3.5, 4, 4.5, 5, 7, and 10 min) after the sample was placed into the dispersion medium. This method is known as “kinetic dispersion mode” as it allows for the evaluation of the aqueous dispersion behavior of a powdered sample, especially the spontaneous dispersion behavior into single MRPs. The sample equivalent of 2 mg MRP per 10 mL dispersion medium was used.
Crystalline AnalysisThe crystalline properties of the MRPs and G-MRPs were evaluated by X-ray powder diffraction (XRPD). The XRPD analysis was conducted using a Geiger-Flex diffractometer (SmartLab; Rigaku Corp., Tokyo, Japan) with CuKα1 radiation and a Ni filter at 40 kV and using a current of 30 mA. The samples were scanned over 5° to 45° of the 2θ range at a scan speed of 5°/min.
Differential Scanning Calorimetry (DSC)DSC curves were obtained using DSC instruments (DSC-60, Shimadzu Corp., Kyoto, Japan). Five milligrams of sample powder was placed in an aluminum pan, which was then crimped. The sample was heated at fixed rate 5°C/min from room temperature to 160°C higher than the melting point of the material, while being purged with dry nitrogen gas.
Contact Angle MeasurementFifty milligrams of various G-MRPs were filled into an 8 mmϕ die. By using a universal compression-tensile testing machine (Autograph AG-5000D, Shimadzu Corp.), the samples were compressed by punches at a compression rate of 1 mm/min at a compression pressure of 1500 kgf to prepare a tablet with a 1.65 mm thickness. The contact angle was measured using an automatic contact angle instrument (DMS-400, Kyowa Interface Science). Purified water was placed in a micro-syringe at room temperature, and a 1 µL water droplet at the needle tip was transferred to the surface of the tablet to form a water lens. The height (H) and diametrical length (L) of the lens were measured immediately after formation of the droplet. The contact angle (θ) was calculated based on the following equation.
H: height of the droplet.
L: diametrical length of the contact lens.
Results and Discussion
Concepts of the Spray-Drying and Drop Freeze-Drying ProcessesMRPs were successfully prepared through the spray-drying method process in a short amount of time, but the resultant powder exhibited poor handling properties with static electricity. The powder also highly aggregated in water and exhibited poor dispersibility. Therefore, to disperse the MRPs into water as primary particles and to dry the particles to make dry powders while maintaining the mono-dispersed state, the MRPs were dispersed in an aqueous solution of the additives and immediately frozen to separately fix each particle in ice. Furthermore, the iced particles were freeze-dried (sublimated) to eliminate the ice as solid state without allowing the ice to liquefy and to precipitate the additives in the ice. This was performed to allow the MRPs to become fixed while mono-dispersed in the matrix of the carrier. Therefore, instead of freezing the entire solution in a vial, as is typical in the production process of conventional injectable products, the freeze-dried product favored transformation into spherical granules because the solution was frozen as separate droplets.
An encapsulator was used to quickly prepare droplets of uniform size and shape. The encapsulator was originally developed to prepare seamless microcapsules via the drying-in-liquid method.17) The droplet particle size diameter can be easily controlled by adjusting the aperture size of the nozzle, the feed rate, and the vibration frequency. Other features are also available that allow for the rapid preparation of droplets at several hundreds or thousands per second with a sharp PSD. Spherical microcapsules can be prepared as the membrane material solidifies at the interface when the droplets are dripped into a coagulation reaction solution. In this study, an aqueous suspension containing the MRPs was employed as the dripping liquid, and liquid nitrogen was used as the dispersion medium to rapidly obtain ice beads with a uniform particle size. Subsequently, the generated ice beads were then freeze-dried. In a typical lyophilization process, a cake-like freeze-dried product can be obtained by vacuum drying (sublimation) of the ice after the drug is dissolved in an aqueous solution and the resultant aqueous fluid is filled into a vial and frozen. In contrast, when the solution is dripped stepwise using an encapsulator, the ice instantly forms beads by maintaining the form of the droplets. A major difference is that formed droplets are dried and solidified to maintain their shape, and thus the obtained lyophilized product transforms into spherical granules with uniform particle diameters. Using the dropping lyophilization method described above, the dropping and freeze-drying procedures were successfully performed regardless of the type of additive used in the formulation. As Shimizu et al. reported for Leuprolerin,18) in the manufacturing process of PLGA composites, a suspension of PLGA microparticles was dispersed into mannitol solution and then freeze-dried to obtain a bulky freeze-dried product, which formed a cake-like powdery block. After the freeze-dried powder that has been crushed, it was used to fill the vials or syringes. On the contrary, the current G-MRPs would not need to be crushed, as the freeze-dried solid products were obtained in granular shapes with high micromeritic property. With such characteristics, automatic filling into the pre-filled syringe can be expected. Low levels of residual solvent of the G-MRPs are expected because solvent removal by freeze-drying frozen droplets is easier than solvent removal from a bulk solution in a tray. Although dichloromethane measurements are not performed in this study, it is well known that dichloromethane is also used in the W/O/W emulsification process for the Leuprorelin formulation. It has been reported that the organic solvent can be removed almost completely via water vaporization,19) and Leurprorelin products are currently marketed using this manufacturing method. Therefore, removal of the solvent down to the degree of residual tolerance in commercial products is considered as feasible. In addition there was no ARP leakage from MRPs during the spray-drying and drop freeze-drying process because ARP is a poorly water-soluble drug. In fact, the actual drug content measured in the G-MRP 50%MNT was 36.4%, which was equivalent to the theoretical value (=36.36%).
Observations of Particle Morphology and Surface Cross-SectionsThe appearances of the ARP bulk particles, MRPs and G-MRPs were observed by SEM (Fig. 2). The MRPs were spherical particles of 5 to 20 µm in size with a smooth surface (Fig. 2B), which is the typical appearance obtained via the general spray-drying method. In the MRPs, the ARP crystals (Fig. 2A) had completely disappeared, suggesting that the ARP drug had become completely embedded in the polymer matrix of the PLGA. Although all G-MRPs were round granules of approximately 400 µm in size, differences in their appearances were observed depending on the composition and the amount of carrier. In particular, G-MRP 50%MNT formulation, which was mainly composed of mannitol, had a spherical shape due to rapid crystallization in the droplets. Both the G-MRP 20%HPC and G-MRP 50%HPC (not shown) formulations exhibited dense fibrous network structures, but some breakage of the granules was observed in the 20%HPC, which contained a lesser amount of carrier (Fig. 2C1). Although differences in surfaces of each G-MRP is difficult to confirm from the overall SEM images (Figs. 2C1, D1, E1, F1), the highly magnified SEM images clearly revealed that small PLGA particles were exposed on the surface of only the G-MRP 20%HPC granules (Fig. 2C2). On the other hand, the G-MRP 50%PVA, G-MRP 20%PVA, and G-MRP 50%MNT formulations differed from the G-MRPs in the HPC carrier in that small PLGA microparticles were not observed on the surface of the granules (Figs. 2D2, E2, F2). Particularly, it was found that the outside of the particles was covered with a smooth film-like outer shell (Fig. 2F2). These finding suggests that both G-MRP PVA and G-MRP MNT formulations, which both contained PVA, had thin shell structures on the surface of the granules. In contrast, the cross-sectional images indicate that small MRPs were embedded in the fibrous network structures of all of the G-MRPs (Figs. 2C3, D3, E3, F3). From these observations, it was confirmed that all the G-MRPs, regardless of the carrier used in the formulations (HPC, PVA, or MNT), exhibited the particle-in-granule structure by which PLGA microparticles were embedded in the granules within the porous matrix of the carrier. The size distributions and representative particle diameters of the G-MRPs shown in Fig. 3 and Table 4 indicate that all of the MRP granules had identical sizes ranging from 100 to 1000 µm and the same average particle diameter (D50) of approximately 350 µm. This value is equivalent to the nozzle diameter used during the dropping process (Fig. 3). In this study, by unifying the driving conditions in the droplet formation process using the encapsulator, a homogeneous distribution with a uniform particle size was obtained as a result of the formation of the size-controlled droplets, even among the different formulations that were employed. This suggests that it is possible to freely adjust the particle size of the granules by changing the formation conditions of the droplet. As the droplets are immediately frozen in liquid nitrogen, the shape and size of the droplets are retained as they form ice particles. Therefore, G-MRPs are small spherical particles derived from the droplets and have the morphological characteristics of a lyophilized cake-like porous internal structure. The image analysis revealed that the small MRPs had an average particle diameter of 10.6 µm (D50). This value was identical to that obtained from the SEM images of the MRPs embedded in each type of G-MRP granule.
Table 4 Representative PSDs of the MRPs and G-MRPs Composed of Various Ratios of MRPs and Additives
| D10 (µm) | D50 (µm) | D90 (µm) |
|---|
| MRP (image analysis) | 4.08 | 10.6 | 25.5 |
| G-MRP 50%HPC | 197 | 350 | 658 |
| G-MRP 20%HPC | 193 | 346 | 655 |
| G-MRP 50%PVA | 186 | 343 | 657 |
| G-MRP 20%PVA | 200 | 355 | 663 |
| G-MRP 50%MNT | 209 | 361 | 665 |
The data were measured via laser diffraction (dry condition).
To clarify the dispersion behaviors of the MRPs and the G-MRPs in the injection fluid, the transitions of the PSDs were traced after the samples were placed in the aqueous phase. The powder samples were directly placed in a detection cell filled with the dispersion medium and attached to a laser diffractometer. After mixing with a stirrer, the diffraction intensity value of the dispersed fluid was continuously measured for 10 min, and the transitions of the D50 value obtained from the PSDs were plotted (Fig. 4). As a control, the geometrical D50 value (geo D50) of the primary MRPs, i.e., 10.6 µm, which represents the average size that was visually observed in the morphologically analyzed SEM images of the MRPs, was drawn as a dotted line in the figure. Immediately after the MRPs were placed in water, the D50 values increased by 30-fold compared to the geo D50 value and did not decrease even after 10 min of stirring. This result demonstrates that individual MRPs became highly aggregated in water, and de-aggregation did not occur even while stirring. In contrast, each G-MRP formulation exhibited different kinetic dispersion behaviors. The G-MRP 50%HPC and G-MRP 50%PVA formulations yielded D50 values that exceeded 100 µm when placed in water, and then gradually exhibited dispersion behaviors over several minutes of stirring. In both samples that contained high amounts of HPC or PVA, it was assumed that, at the beginning of the dispersion process, the concentrated phase dissolved the water soluble polymer, thus maintaining a high viscosity and preventing dispersion of the MRPs. As the phase was gradually diluted with water and the viscosity decreased, the aggregates dissociated into small particles. However, despite the dissociation with dilution, the D50 value did not decrease to the value of the primary MRP particle diameter, and therefore, complete dispersion was not attained. In contrast, the G-MRP 20%HPC and G-MRP 20%PVA samples that contained less polymer exhibited D50 values of approximately 30 µm immediately after the dispersion, and their initial dispersibility was better compared with the 50% samples. Such favorable kinetic dispersibility likely occurred because a highly viscous polymeric phase did not form. The D50 values of both the 20%HPC and 20%PVA formulations did not decrease to the size of the visually observed primary particle diameter (10.6 µm) even after 10 min, suggesting that a portion of the MRPs remained weakly aggregated. The small mass ratio of the additives was thought to lead to a relative increase in the number of MRPs per unit mass of the matrix, and thus, the MRPs became dried (solidified) without being perfectly dispersed as a primary particle in the matrix base of the G-MRPs.
Therefore, the proportion of additives was increased to up to 50%, and D-mannitol was used as a primary additive to prevent an increase in the viscosity upon dissolving the polymer, thus leading to the G-MRP 50%MNT formulation containing less PVA. One minute after the sample was placed into the dispersion water, a D50 value of 14.6 µm was observed, indicating that the dispersion state successfully approached a monodispersion of MRP primary particles. Such excellent dispersion features of D-mannitol, which quickly dissolves in water, lead to a low viscosity and thus facilitating the dispersion of each MRP. Carrio et al. reported that PVA molecules adsorb onto the PLGA particles in a linear chain, which induces a steric barrier effect that inhibits the approach of MRPs.20) The small amount of PVA included in the G-MRP formulation was thought to actively promote the dispersion of the MRPs to inhibit re-aggregation in the G-MRPs, which could represent one of the factors that led to favorable dispersibility. In addition, formulations with mass ratios that corresponded to an equivalent amount of additives and MRPs also moderately facilitated the dispersion of the MRPs in the matrix of the G-MRPs.
In clinical practice, it is highly desirable to medical practitioners that injections are able to be administered in a short period of time after addition of the dispersion fluids, such as water for injections that are manufactured as powdered products. Therefore, the PSDs of the dispersed particles at 1 min after hydration are plotted in Fig. 5. The G-MRP 20%HPC, G-MRP 20%PVA, and GMRP 50%MNT formulations were selected due to their favorable dispersibility within a short time. The distribution of primary MRPs obtained from the image analysis is also shown as a control. Their representative particle diameters are shown in Table 5. Figure 5 indicates that the size distributions of the dispersed MRPs in the G-MRPs were not consistent with visually obtained geometric PSD of individual MRPs, suggesting that the MRPs did not achieve complete primary dispersion in the water. The MRPs themselves exhibited a bimodal distribution with peaks at approximately 50 and 400 µm, which revealed that the PLGA particles have a high tendency to aggregate. The maximum particle diameter obtained from the distribution curve exceeded 1000 µm, indicating a risk of clogging a 26G subcutaneous needle with a 450 µm aperture diameter, which are generally used for injecting PLGA formulations. The G-MRP 20%HPC and G-MRP 20%PVA formulations had D50 values of 23.7 and 30.6 µm, respectively. Although some improvements in the MRPs were recognized, the maximum D50 values were 91.1 µm for the G-MRP 20%HPC and 152 µm for the G-MRP 20%PVA at 1 min, which correspond to approximately one-fifth to one-third of a pore diameter, raising concerns of possible needle clogging at the time of injection. In contrast, the G-MRP 50%MNT formulation exhibit a D50 value of 14.6 µm and a maximum value of 46.1 µm, indicating excellent dispersibility and a markedly reduced risk of clogging the injection needle. In addition, considering usability in medical practice, the stability of the dispersed particles was also evaluated during storage after preparation. The G-MRP 50%MNT formulation that exhibited the best dispersion behavior when placed in an injection syringe filled with the injection fluid. The PSD of this formulation was measured after 1, 5, 10, 30, and 50 min. It was found that the PSD just after dispersion remained unchanged after storage for 50 min, and the original dispersion state was maintained (Fig. 6). These results revealed that, for the G-MRP 50%MNT formulation, the MRPs immediately formed a near perfect dispersion as primary particles in water and maintained a favorable dispersion state without re-aggregating during long-term storage. Thus, this optimum formulation demonstrated excellent characteristics for use in ready-to-use depot injectables.
Table 5. Representative Particle Sizes of the MRPs and G-MRPs Composed of Various Ratios of MRPs and Additives
| D10 (µm) | D50 (µm) | D90 (µm) | Maximum (µm) |
|---|
| MRP | 36.7 | 385 | 715 | >1000 |
| G-MRP 20%HPC | 9.9 | 23.7 | 47.4 | 91.1 |
| G-MRP 20%PVA | 11.4 | 30.6 | 72.7 | 152 |
| G-MRP 50%MNT | 7.32 | 14.6 | 27.1 | 46.1 |
| MRP (Image analysis) | 4.08 | 10.6 | 25.5 | 15.0 |
The data were measured via laser diffraction (wet condition).
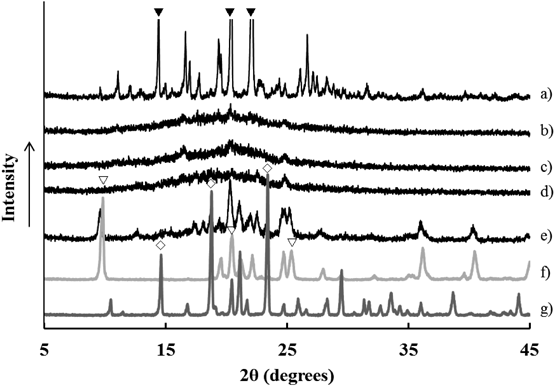
The crystalline state of the G-MPRs was evaluated by XRPD. The XRPD patterns of various G-MRPs, intact ARP crystals and MPRs are shown in Fig. 7. The intact ARP crystals showed characteristic diffraction peaks at 14.5, 18.7, and 23.3° of 2θ and was confirmed to be in the metastable III crystalline form.21) As previously reported for spray-dried PLGA products by Apoorva et al., the XRPD pattern of the PLGA microparticles exhibited no characteristic peaks of ARP but displayed a halo pattern, suggesting that ARP was present in an amorphous state in the particles.22) Whereas, for the G-MRP 50%HPC (data not shown), G-MRP 20%HPC, G-MRP 50%PVA (data not shown), and even G-MRP 20%PVA formulations, similar halo patterns were observed, indicating that APR was not crystallized during the drop freeze-drying process. On the other hand, for the G-MRP 50%MNT formulation, specific diffraction peaks at 9.4 and 21.7° derived from crystalline mannitol δ were observed in the 2θ range. The precipitation of metastable crystals that are different from the stable β form of original mannitol was previously reported to occur during crystallization at 0°C by Yoshinari et al.,23,24) and our manufacturing process was consistent with this report.
The thermal behavior profiles of intact ARP, intact PLGA, MRPs, and the G-MRP 50%MNT formulation measured by differential scanning calorimetry (DSC) are shown in Fig. 8. It was verified that ARP was in the III crystalline form, (a) as ARP showed an endothermic onset peak at 138.5°C based on melting.21) The PLGA bulk powder exhibited an endothermic onset peak at 46.7°C, which was assigned as the glass transition temperature (Tg) (c) as indicated by the supplier. The Tg did not change even after the physically mixing with ARP (b). In the MRP products in which the ARP and PLGA had been co-spray-dried, the Tg of PLGA rose to 50.2°C from an original onset peak (46.7°C), suggesting that ARP was interacted with the PLGA at the molecular level and it distributed as an amorphous state in the PLGA molecular chain (d). In the G-MRP 50%MNT formulation, PLGA had nearly the same Tg as MRP (e), indicating that the state of ARP did not change even after lyophilization with additives. It was presumed that the increase in the inherent Tg of the PLGA after treatment would contribute to improving the thermodynamic stability of the formulation during storage. In both DSC charts (d) and (e) of MRP and G-MRP 50%MNT, the broad exothermic peak and the sharp endothermic peak around 135–140°C were observed, corresponding to re-crystallization and melting of ARP, respectively. In addition, G-MRP 50%MNT had another endothermic peak over 150°C derived from melting of mannitol.
Rapid Dispersion Process and the Mechanism of the G-MRPsThe difference in the dispersion behavior between the G-MRPs in the PVA formulation and the MNT formulation was evaluated based on their wettability by water. The contact angle of various samples of MRP, G-MRP and additives, namely PVA bulk and MNT bulk, were measured by placing an aqueous drop of the sample on the compressed surface (Fig. 9). Each G-MRP sample had lower contact angles than that of the MRPs. In addition, the G-MRP 50%PVA formulation that contained a higher PVA content had a lower contact angle than the G-MRP 20%PVA formulation. The high wettability of 50% of the PVA formulation could not accelerate the dispersion behavior of individual PLGA particle due to the elevated viscosity of the penetrating water into the granule, as described above. The G-MRPs in the G-MRP 50%MNT formulation exhibited the lowest contact angle in which the water-soluble D-mannitol was formulated as a carrier. It was assumed that the high wettability is the factor that promoted the water penetration into the internal granules and the improved dispersibility of the individual MRPs.
As revealed by these results, excellent dispersion of the G-MRP 50%MNT formulation was achieved by the following process (schematic diagram shown in Fig. 10). Small MRPs are uniformly dispersed in the porous matrix of the granules composed of D-mannitol as the main carrier. When dispersed in water, water rapidly penetrates the matrix pores of the granules, D-mannitol is immediately dissolved, and the MRPs are released as free particles. PVA molecular chains can adsorb on the surface of the MRPs for steric stabilization and the prevention of inter-particle aggregation, thus leading to the enhanced dispersibility of the MRPs in the aqueous medium.
Conclusion
In this study, spherical granules containing sustained-release PLGA microparticles (G-MRPs) were successfully prepared via a two-step preparation method composed of spray-drying and drop freeze-drying processes. Small MRPs were observed to be embedded inside of all G-MRPs, which exhibited the specific microparticles-in-granule structure. PLGA particles directly supplied from the spray-drying process strongly assembled to one another in the aqueous phase, forming large aggregations and poor dispersibility, whereas the G-MRPs immediately dispersed in the aqueous phase and exhibited potential excellent dispersibility in injection fluids. The physicochemical evaluation of MRPs and G-MRPs suggested that the drug and PLGA were molecularly interacted, leading to improved thermal stability of the MRPs. Their interaction did not change even through the drop freeze-drying process used to prepare the G-MRP. Based on the evaluation of the contact angles of the MRPs and G-MRPs, it was confirmed that the G-MRPs were more wettable than the drug and MRPs, resulting in excellent dispersibility in the aqueous phase. Such characteristics of the G-MRPs are attributed to the rapid dissolution of water-soluble D-mannnitol and the steric hindrance effect of PVA molecular chains. According to the outcomes of the current study, the G-MRPs exhibited favorable properties for depot-type sustained-release suspension injectable formulations. In addition, the current production process consisting of a two-step method is easier to carry out than the conventional preparation of PLGA products. The method resulted in spherical granular formulations with well-controlled particle sizes that can be used for the automatic filling of pre-filled syringes.
Conflict of Interest
The authors declare no conflict of interest.
References
- 1) Chen T., Miller T. F., Prasad P., Lee J., Krauss J., Miscik K., Kalafsky G., McLeod J. F., J. Clin. Pharmacol., 40, 475–481 (2000).
- 2) Jones A. J., Putney S., Johnson O. L., Cleland J. L., Adv. Drug Deliv. Rev., 28, 71–84 (1997).
- 3) Ogawa Y., Yamamoto M., Okada H., Yashiki T., Shimamoto T., Chem. Pharm. Bull., 36, 1095–1103 (1988).
- 4) Okada H., Heya T., Ogawa Y., Toguchi H., Shimamoto T., Pharm. Res., 8, 584–587 (1991).
- 5) Okada H., Doken Y., Ogawa Y., Toguchi H., Pharm. Res., 11, 1143–1147 (1994).
- 6) Okada H., Doken Y., Ogawa Y., Toguchi H., Pharm. Res., 11, 1199–1203 (1994).
- 7) Rajni S., Nicholas S., Clin. Interv. Aging, 4, 259–267 (2009).
- 8) Tunn U. W., BMC Urol., 11, 15 (2011).
- 9) Kawamura K., PDA Journal of GMP and Validation in Japan, 8, 2–17 (2006).
- 10) Paolo G., Bice C., Ida G., Ubaldo C., Giovanni P., Drug Dev. Ind. Pharm., 27, 745–750 (2001).
- 11) Broadhead J., Edmond Rouan S. K., Rhodes C. T., Drug Dev. Ind. Pharm., 18, 1169–1206 (1992).
- 12) Clarke N., O’Connor K., Ramtoola Z., Drug Dev. Ind. Pharm., 24, 169–174 (1998).
- 13) Mortini M., Pedroncelli A., Tengattini F., Pagani M., Gianola D., Cortesi L., Pagani G., Lancranijan I., “Pharmaceutical Particulate Carries, Therapeutic Applications,” Marcel Dekker, NY, 1993, pp. 227–274.
- 14) Okada H., Adv. Drug Deliv. Rev., 28, 43–70 (1997).
- 15) Burris K. D., Molski T. F., Xu C., Ryan E., Tottori K., Kikuchi T., Yocca F. D., Molinoff P. B., J. Pharmacol. Exp. Ther., 302, 381–389 (2002).
- 16) Hiraoka S., Uchida S., Namiki N., Chem. Pharm. Bull., 62, 654–660 (2014).
- 17) De Prisco A., Maresca D., Ongeng D., Mauriello G., LWT—Food Sci. Technol., 61, 1–11 (2014).
- 18) Shimizu H., Nonomura M., Futo M., Mukai K., WO 9937288 (1999).
- 19) Okada H., Doken Y., Yamada A., Japan Patent H9-221417 (1997).
- 20) Carrio A., Schwach G., Coudane J., Vert M., J. Control. Release, 37, 113–121 (1995).
- 21) Braun D. E., Gelbrich T., Kahlenberg V., Tessadri R., Wieser J., Griesser U. J., J. Pharm. Sci., 98, 2010–2026 (2009).
- 22) Apoorva P., Jairam M., Rajesh K., Dipak K. M., Pharm. Dev. Technol., 21, 43–53 (2016).
- 23) Yoshinari T., Forbes R. T., York P., Kawashima Y., Int. J. Pharm., 247, 69–77 (2002).
- 24) Yoshinari T., Forbes R. T., York P., Kawashima Y., Int. J. Pharm., 258, 109–120 (2003).