Experimental
ChemistryMelting points (mp) were determined on Griffin apparatus, and the values given are uncorrected. IR spectra were determined on Shimadzu IR 435 spectrophotometer (KBr, cm−1). 1H-NMR spectra were carried out using Varian Mercury-300 (300 or 400 MHz) Spectrophotometer using tetramethylsilane (TMS) as internal standard. Chemical shift values are recorded in ppm on δ scale, Micro Analytical Center, Cairo University, Egypt and Micro Analytical Unit, Faculty of Pharmacy, Cairo University, Egypt. Coupling patterns are described as follows: s, singlet, d, doublet, t, triplet, m, multiplet. J describes a coupling constant. Mass spectra were recorded on a GCMP-QP1000 EX Mass spectrometer, Micro Analytical Center, Cairo University, Egypt. Elemental analyses were carried out at the Micro Analytical Center, Al-Azhar University, Egypt. Progress of the reactions was monitored using TLC sheets pre-coated with UV fluorescent silica gel Merck 60F 254, using toluene–ethanol (4.5 : 0.5) and were visualized using UV lamp. All chemicals were obtained from Aldrich, Fluka, or Merck chemicals. N-Carbamimidoyl-4-[(6-chloropyridazin-3-yl)amino]benzensulfonamide (1) was prepared as reported.11)
General Procedure for the Preparation of 2a–fAn equimolar amount of 1 (3.265 g, 0.01 mol) and the appropriate amine (0.01 mol) in 20 mL n-butanol was heated under reflux for 15 h. The reaction mixture was cooled, and the separated solid was filtered, dried, and recrystallized from ethanol.
N-Carbamimidoyl-4-[(6-phenylaminopyridazin-3-yl)amino]benzensulfonamide (2a)Yield: 1.63 g (42.6%); mp: 190–192°C; 1H-NMR (dimethyl sulfoxide (DMSO)-d6, 300 MHz) δ: 6.70 (2H, s, D2O exchangeable), 7.36 (1H, d, J=8.40 Hz), 7.11–7.76 (9H, m), 7.57 (1H, d, J=8.40 Hz), 9.80 (1H, s, D2O exchangeable), 10.03 (1H, s, D2O exchangeable). 13C-NMR (DMSO-d6, 100 MHz) δ: 113.43, 121.17, 122.35, 126.77, 127.81, 129.77, 130.07, 131.46, 134.71, 151.13, 158.10. IR (KBr) cm−1: 3435, 3338, 3267, 3184, 3132, 1330 and 1138. MS m/z: 385.00 [M+2]+, 384 [M+1]+, 383.00 (M+), 291.00, 234.00. Anal. Calcd for C17H17N7O2S (383.43): C, 53.25; H, 4.47; N, 25.57; Found: C, 53.31; H, 4.49; N, 25.71.
N-Carbamimidoyl-4-[(6-(4-methylphenyl)aminopyridazin-3-yl)amino]benzensulfonamide (2b)Yield: 0.78 g (19.6%); mp: 130–132°C; 1H-NMR (DMSO-d6, 400 MHz,) δ: 2.35 (3H, s), 6.59 (2H, d, J=8.56 Hz), 6.73 (2H, s, D2O exchangeable), 7.22 (2H, d, J=8.04 Hz), 7.43 (2H, d, J=8.60 Hz), 7.56 (1H, s, D2O exchangeable), 7.73 (1H, d, J=8.76 Hz), 7.80 (2H, d, J=8.72 Hz), 7.87 (1H, d, J=8.76 Hz), 9.55 (1H, s, D2O exchangeable), 9.76 (1H, s, D2O exchangeable). 13C-NMR (DMSO-d6, 100 MHz) δ: 20.98, 113.13, 116.66, 117.91, 121.41, 127.25, 127.74, 130.19, 133.39, 137.40, 150.51, 158.50. IR (KBr) cm−1: 3452, 3433, 3371, 3332, 3215, 2850–2900, 1332 and 1130. MS m/z: 397.00 (M+), 233.00. Anal. Calcd for C18H19N7O2S (397.45): C, 54.39; H, 4.82; N, 24.67; Found: C, 54.47; H, 4.91; N, 24.83.
N-Carbamimidoyl-4-[(6-(2-fluorophenyl)aminopyridazin-3-yl)amino]benzensulfonamide (2c)Yield: 1.56 g (39.0%); mp: 196–198°C; 1H-NMR (DMSO-d6, 300 MHz,) δ: 5.65 (2H, s, D2O exchangeable), 6.53 (2H, d, J=6.60 Hz), 6.54 (2H, d, J=6.60 Hz), 6.55 (1H, d, J=8.40 Hz), 6.45–7.79 (4H, m), 7.36 (1H, d, J=8.40 Hz), 9.80 (1H, s, D2O exchangeable). 13C-NMR (DMSO-d6, 100 MHz) δ: 110.76, 112.79, 115.68, 118.21, 121.29, 124.60, 127.23, 127.70, 130.03, 131.28, 147.99, 151.83, 158.22. IR (KBr) cm−1: 3454, 3433, 3371, 3331, 3219, 1303 and 1128. MS m/z: 401.00 (M+), 400.00 [M−1]+, 291.00 233.00. Anal. Calcd for C17H16FN7O2S (401.42): C, 50.87; H, 4.02; N, 24.43; Found: C, 50.96; H, 4.08; N, 24.55.
N-Carbamimidoyl-4-[(6-(2-chloropyridin-3-ylamino)pyridazin-3-yl)amino]benzensulfonamide (2d)Yield: 1.74 g (41.7%); mp: 184–186°C; 1H-NMR (DMSO-d6, 300 MHz) δ: 5.65 (2H, s, D2O exchangeable), 6.53 (2H, d, J=6.60 Hz), 6.54 (1H, d, J=8.40 Hz), 6.55–7.79 (3H, m), 7.37 (2H, d, J=6.60 Hz), 7.38 (1H, d, J=8.40 Hz), 9.80 (1H, s, D2O exchangeable). 13C-NMR (DMSO-d6, 100 MHz) δ: 112.81, 118.20, 121.34, 127.22, 127.71, 130.05, 131.28, 137.56, 143.15, 148.58, 151.80, 158.22. IR (KBr) cm−1: 3489, 3435, 3417, 3398, 3342, 3223, 1309 and 1134. MS m/z: 420.10 [M+2]+, 418.10 (M+), 416.10 [M−2]+, 290.10. Anal. Calcd for C16H15ClN8O2S (418.86): C, 45.88; H, 3.61; N, 26.75; Found: C, 45.96; H, 3.67; N, 27.02.
N-Carbamimidoyl-4-[(6-(2, 6-dichlorophenyl)aminopyridazin-3-yl)amino]benzensulfonamide (2e)Yield: 0.65 g (14.4%); mp: 260–262°C; 1H-NMR (DMSO-d6, 400 MHz) δ: 6.53 (2H, d, J=8.64 Hz), 6.66 (2H, s, D2O exchangeable), 7.22–7.69 (3H, m), 7.37 (2H, d, J=8.64 Hz), 7.70 (1H, d, J=8.80 Hz), 7.81 (1H, d, J=8.80 Hz), 9.49 (1H, s, D2O exchangeable), 9.84 (1H, s, D2O exchangeable). 13C-NMR (DMSO-d6, 100 MHz) δ: 112.89, 118.20, 121.33, 127.22, 127.72, 130.05, 137.56, 143.14, 148.58, 157.01, 158.48.IR (KBr) cm−1: 3431, 3329, 3234, 3203, 3169, 1253 and 1138. MS m/z: 456.00 [M+4]+, 454.00 [M+2]+, 452.00 (M+), 292.00, 233.00. Anal. Calcd for C17H15Cl2N7O2S (452.32): C, 45.14; H, 3.34; N, 21.68; Found: C, 45.16; H, 3.37; N, 21.74.
N-Carbamimidoyl-4-[(6-2-bromophenylaminopyridazin-3-yl)amino]benzensulfonamide (2f)Yield: 0.60 g (13.0%); mp: >300°C; 1H-NMR (DMSO-d6, 300 MHz,) δ: 6.57 (2H, s, D2O exchangeable), 6.65–7.82 (8H, m), 7.26 (1H, d, J=9.30 Hz), 7.62 (1H, d, J=9.30 Hz), 7.83 (1H, s, D2O exchangeable); 9.57 (1H, s, D2O exchangeable), 9.83 (1H, s, D2O exchangeable). 13C-NMR (DMSO-d6, 100 MHz) δ: 113.85, 118.20, 121.37, 127.22, 127.90, 131.56, 133.08, 135.23, 143.22, 148.58, 150.65, 157.91, 158.45. IR (KBr) cm−1: 3421, 3350, 3329, 3232, 3203, 3170, 1253 and 1138. MS m/z: 463.00 [M+2]+, 461.00 (M+), 460.00 [M−1]+, 234.00. Anal. Calcd for C17H16BrN7O2S (462.32): C, 44.16; H, 3.49; N, 21.21; Found: C, 44.20; H, 3.53; N, 21.29.
General Procedure for the Preparation of (3a, b)A mixture of 1 (3.265 g, 0.01 mol) and the appropriate phenylenediamine (1.08 g, 0.01 mol) in 30 mL absolute ethanol was heated under reflux for 12 h. The reaction mixture was concentrated under reduced pressure, cooled and the formed solid was filtered, dried and recrystallized from ethanol.
N-Carbamimidoyl-4-[(6-(3-aminophenylamino)pyridazin-3-yl)amino]benzensulfonamide (3a)Yield: 2.93 g (73.8%); mp: 130–132°C; 1H-NMR (DMSO-d6, 400 MHz,) δ: 6.64–7.81 (4H, m), 6.67 (1H, d, J=8.56 Hz), 6.79 (2H, s, D2O exchangeable), 6.91 (2H, d, J=7.08 Hz), 7.36 (2H, d, J=7.52 Hz), 7.46 (1H, d, J=8.60 Hz), 7.62 (1H, s, D2O exchangeable), 10.45 (1H, s, D2O exchangeable), 10.52 (1H, s, D2O exchangeable). 13C-NMR (DMSO-d6, 100 MHz) δ: 111.86, 113.58, 116.17, 127.27, 127.84, 130.50, 131.50, 133.07, 135.23, 150.32, 150.96, 158.07, 158.52. IR (KBr) cm−1: 3435, 3400, 3344, 3215, 1226 and 1130. MS m/z: 399.00 [M+1]+, 398.00 (M+), 292.00, 233.00. Anal. Calcd for C17H18N8O2S (398.44): C, 51.25; H, 4.55; N, 28.12; Found: C, 51.41; H, 4.59; N, 28.34.
N-Carbamimidoyl-4-[(6-(4-aminophenylamino)pyridazin-3-yl)amino]benzensulfonamide (3b)Yield: 3.48 g (87.6%); mp: 96–98°C; 1H-NMR (DMSO-d6, 300 MHz,) δ: 5.52 (2H, s, D2O exchangeable), 6.52 (2H, d, J=6.60 Hz), 6.53 (2H, d, J=6.60 Hz), 6.63 (1H, d, J=8.70 Hz), 7.28 (1H, d, J=8.70 Hz), 7.36 (2H, d, J=6.90 Hz), 7.37 (2H, d, J=6.90 Hz), 8.03 (1H,s, D2O exchangeable), 9.00 (1H, s, D2O exchangeable), 9.80 (1H, s, D2O exchangeable). 13C-NMR (DMSO-d6, 100 MHz) δ: 110.02, 112.92, 114.89, 116.45, 118.31, 127.72, 128.15, 131.32, 141.40, 151.69, 158.22. IR (KBr) cm−1: 3489, 3435, 3414, 3398, 3342, 3219, 1232 and 1132. MS m/z: 398.10 (M+), 397.10 [M−1]+, 291.10, 233.00. Anal. Calcd for C17H18N8O2S (398.44): C, 51.25; H, 4.55; N, 28.12; Found: C, 51.37; H, 4.58; N, 28.27.
General Procedure for the Preparation of (4a, b)An equimolar amount of 3a (3.98 g, 0.01 mol) and the selected acid chloride (0.01 mol) in 25 mL absolute ethanol was heated under reflux for 9 h. The reaction mixture was concentrated under reduced pressure and the formed solid was filtered dried and recrystallized from ethanol.
N-(3-(6-(4-(N-Carbamimidoylsulfamoyl)phenylamino)pyridazin-3-ylamino)phenyl)acetamide (4a)Yield: 0.59 g (13.4%); mp: 196–198°C; 1H-NMR (DMSO-d6, 300 MHz) δ: 2.02 (3H, s), 6.29 (2H, s, D2O exchangeable), 6.56 (1H, d, J=8.70 Hz), 6.62 (2H, d, J=6.90 Hz), 6.63 (2H, d, J=6.90 Hz), 6.66–7.74 (4H, m), 7.39 (1H, d, J=8.70 Hz), 10.20 (1H, s, D2O exchangeable). 13C-NMR (DMSO-d6, 100 MHz) δ: 40.96, 114.92, 117.15, 119.25, 123.01, 127.42, 128.45, 129.22, 131.17, 132.72, 136.49, 146.81, 147.95, 157.03, 158.68. IR (KBr) cm−1: 3442, 3394, 3342, 3309, 3224, 3209, 3140, 2922–2854, 1647, 1244 and 1136. MS m/z: 440.20 (M+), 439.20 [M−1]+, 291.10, 233.10. Anal. Calcd for C19H20N8O3S (440.48): C, 51.81; H, 4.58; N, 25.44; Found: C, 51.89; H, 4.56; N, 25.71.
N-(3-(6-(4-(N-Carbamimidoylsulfamoyl)phenylamino)pyridazin-3-ylamino)phenyl)-3-chloropropanamide (4b)Yield: 0.70 g (14.3%); mp: 150–152°C; 1H-NMR (DMSO-d6, 300 MHz,) δ: 2.83 (2H, t, J=6.30 Hz), 3.89 (2H, t, J=6.30 Hz), 6.33 (2H, s, D2O exchangeable), 6.27–7.95 (8H, m), 6.61 (1H, d, J=8.40 Hz), 7.43 (1H, d, J=8.70 Hz), 9.94 (1H, s, D2O exchangeable), 10.08 (1H, s, D2O exchangeable), 10.39 (1H, s, D2O exchangeable). 13C-NMR (DMSO-d6, 100 MHz) δ: 33.61, 55.09, 114.50, 117.23, 119.03, 121.86, 127.13, 127.35, 127.45, 128.01, 130.73, 135.43, 150.41, 157.74, 158.48, 169.74. IR (KBr) cm−1: 3446, 3342, 3315, 3251, 3201, 3151, 2962–2835, 1676, 1313 and 1136. MS m/z: 491.10 [M+3]+, 489.10 [M+1]+, 291.10, 233.00. Anal. Calcd for C20H21ClN8O3S (488.95): C, 49.13; H, 4.33; N, 22.92; Found: C, 49.24; H, 4.39; N, 23.10.
General Procedure for the Preparation of (5a, b)A solution of 3a (3.98 g, 0.01 mol) and the appropriate isothiocyanate (0.01 mol) in 25 mL absolute ethanol was heated under reflux for 10 h. The solution was concentrated under reduced pressure and the residue triturated with ether. The formed solid was filtered, dried and recrystallized from ethanol.
N-Carbamimidoyl-4-(6-(3-(3-ethylthiourea)phenylamino)pyridazin-3-ylamino)benzensulfonamide (5a)Yield: 4.24 g (87.4%); mp: 180–182°C; 1H-NMR (DMSO-d6, 400 MHz,) δ: 1.12 (3H, t, J=7.08 Hz), 3.50 (2H, q, J=7.20 Hz), 6.69 (2H, s, D2O exchangeable), 6.54–7.79 (8H, m), 7.55 (1H, d, J=8.64 Hz), 7.65 (1H, d, J=8.64 Hz), 8.08 (1H, s, D2O exchangeable), 9.58 (1H, s, D2O exchangeable), 9.81 (1H, s, D2O exchangeable). 13C-NMR (DMSO-d6, 100 MHz) δ: 14.45, 65.39, 113.73, 119.00, 121.77, 122.90, 126.64, 127.90, 129.38, 131.48, 139.37, 142.66, 150.81, 157.93, 158.51, 180.36. IR (KBr) cm−1: 3462, 3446, 3433, 3334, 3292, 3246, 3213, 3149, 3109, 2972–2929, 1338 and 1141. MS m/z: 484.90 [M−1]+, 291.90, 233.00. Anal. Calcd for C20H23N9O2S2 (485.59): C, 49.47; H, 4.77; N, 25.96; Found: C, 49.62; H, 4.82; N, 26.13.
N-Carbamimidoyl-4-(6-(3-(3-allylthiourea)phenylamino)pyridazin-3-ylamino)benzensulfonamide (5b)Yield: 2.12 g (42.6%); mp: 82–84°C; 1H-NMR (DMSO-d6, 300 MHz,) δ: 4.12 (2H, t), 5.10–5.34 (2H, m), 5.83–5.94 (1H, m), 6.67 (2H, s, D2O exchangeable), 6.52–7.80 (8H, m), 7.58 (1H, d, J=8.70 Hz), 7.65 (1H, d, J=8.70 Hz), 7.83 (1H, s, D2O exchangeable), 8.16 (1H, s, D2O exchangeable), 9.89 (1H, s, D2O exchangeable), 10.34 (1H, s, D2O exchangeable). 13C-NMR (DMSO-d6, 100 MHz) δ: 46.43, 112.92, 116.18, 116.41, 120.29, 121.82, 126.57, 127.72, 131.32, 134.90, 135.22, 139.55, 142.68, 151.69, 158.23, 158.56, 180.94. IR (KBr) cm−1: 3423, 3369, 3348, 3334, 3217, 2900–2850, 1328 and 1136. MS m/z: 499.00 [M+2]+, 498.00 [M+1]+, 497.00 (M+). Anal. Calcd for C21H23N9O2S2 (497.60): C, 50.69; H, 4.66; N, 25.33; Found: C, 50.78; H, 4.70; N, 25.47.
General Procedure for the Preparation of (6a, b)A mixture of 3b (3.98 g, 0.01 mol) and the appropriate isothiocyanate (0.01 mol) in 25 mL absolute ethanol was heated under reflux for 10 h. The reaction mixture was concentrated under reduced pressure and the residue triturated with ether. The formed solid was filtered, dried and recrystallized from ethanol.
N-Carbamimidoyl-4-(6-(4-(3-ethylthiourea)phenylamino)pyridazin-3-ylamino)benzensulfonamide (6a)Yield: 1.59 g (32.7%); mp: 90–92°C; 1H-NMR (DMSO-d6, 400 MHz) δ: 1.12 (3H, t, J=7.20 Hz), 3.47 (2H, q, J=6.36 Hz), 5.68 (1H, s, D2O exchangeable), 6.52 (2H, d, J=8.56 Hz), 6.68 (2H, s, D2O exchangeable), 7.37 (2H, d, J=8.56 Hz), 7.54 (1H, d, J=8.60 Hz), 7.65 (1H, d, J=8.60 Hz), 7.69 (2H, d, J=8.72 Hz), 7.80 (2H, d, J=8.72 Hz), 8.02 (1H, s, D2O exchangeable), 9.72 (1H, s, D2O exchangeable). 13C-NMR (DMSO-d6, 100 MHz) δ: 14.45, 41.82, 112.82, 118.21, 123.91, 126.64, 127.21, 127.71, 131.26, 139.47, 142.59, 151.82, 158.53, 180.43. IR (KBr) cm−1: 3429, 3334, 3230, 2900–2850, 1334 and 1134. MS m/z: 487.00 [M+2]+, 486.00 [M+1]+, 485.00 (M+), 232.00. Anal. Calcd for C20H23N9O2S2 (485.59): C, 49.47; H, 4.77; N, 25.96; Found: C, 49.55; H, 4.74; N, 26.13.
N-Carbamimidoyl-4-(6-(4-(3-allylthiourea)phenylamino)pyridazin-3-ylamino)benzensulfonamide (6b)Yield: 1.72 g (34.6%); mp: 104–106°C; 1H-NMR (DMSO-d6, 300 MHz) δ: 4.11 (2H, t), 5.07–5.22 (2H, m), 5.65 (2H, s, D2O exchangeable), 5.82–5.93 (1H, m), 6.52 (1H, d, J=8.40 Hz), 6.88 (2H, d),7.35 (2H, d), 7.36 (1H, d, J=8.40 Hz), 7.37 (2H, d), 7.38 (2H, d), 7.83 (1H, s, D2O exchangeable), 9.20 (1H, s, D2O exchangeable), 9.60 (1H, s, D2O exchangeable), 9.80 (1H, s, D2O exchangeable). 13C-NMR (DMSO-d6, 100 MHz) δ: 46.51, 112.82, 116.17, 116.43, 124.01, 126.60, 127.71, 131.28, 134.91, 135.29, 135.98, 151.81, 158.23, 158.53, 181.07. IR (KBr) cm−1: 3452, 3431, 3371, 3331, 3211, 3194, 2999–2856, 1301 and 1130. MS m/z: 498.00 [M+1]+, 497.00 (M+), 232.00. Anal. Calcd for C21H23N9O2S2 (497.60): C, 50.69; H, 4.66; N, 25.33; Found: C, 50.82; H, 4.70; N, 25.39.
Antitumor ActivityMaterials and MethodsIn Vitro AssayThe cytotoxic activity of the tested compounds was measured in vitro against human colon cancer cell line (HCT-116) and breast cancer cell line (MCF-7) in comparison to imatinib, applying Sulforhodamine B (SRB) stain following the method of Skehan et al.10) Human tumor cell lines (MCF-7, HCT-116) used in this study were obtained frozen in liquid nitrogen (−180°C) from the American Type Culture Collection (ATC C, MN, U.S.A.). The tumor cell lines were maintained at the National Cancer Institute, Cairo, Egypt, by serial sub-culturing. Cells were seeded in 96-well microtiter plates at a concentration of (5×104–105cells/well) in a fresh medium and left to attach to the plates for 24 h before treatment of the tested compounds. Test compounds were dissolved in DMSO, and diluted with saline to the appropriate volume. After 24 h, cells were incubated with the appropriate concentration ranges of drugs (0, 5, 12.5, 25 and 50 µg/mL), the wells were diluted to 200 µL with fresh medium and incubation was continued for 48 h at 37°C. Control cells were treated with vehicle alone. Four wells were used for each drug concentration. After 48 h incubation, the cells were fixed with 50 µL of cold 50% trichloroacetic acid for 1 h at 4°C, washed 5 times with distilled water and then stained for 30 min at room temperature with 50 µL 0.4% SRB dissolved in 1% acetic acid. The wells were then washed 4 times with 1% acetic acid. The plates were air dried and the dye was solubilized with 100 µL/well of 10 mM tris base (pH=10.5) for 5 min on a shaker (Orbital shaker OS 20, Boeco, Germany) at 1600 rpm. The optical density (O.D.) of each well was measured spectrophotometrically at 564 nm with an ELISA microplate reader (Meter tech. Σ 960, U.S.A.). The percentage of cell survival was calculated as follows: survival fraction=O.D. (treated cells)/ O.D. (control cells). The relation between surviving fraction and compound concentration was plotted to get the survival curve for tumor cell line after the specified time. The concentration required for 50% inhibition of cell viability (IC50) was calculated for each tested compound (Table 1).
In Vivo AssayInduction of EAC Solid Tumors in MiceFemale Swiss albino mice with original body weight of 20–26 g were used in the current experiment, and fixed housing conditions were maintained at a normal dark/light cycle, mice were housed in groups of twelve in polyethylene cages where food and water were provided ad libitum. The Research Ethics Committee at the Faculty of Pharmacy, Suez Canal University (license number 20148A2), approved the study of protocol. EAC is commonly employed as a solid form18,19) and easy to grow in suspension, when injected in the peritoneal cavity of female mice. Mice carrying EAC cell line were obtained from the Department of Tumor Biology at the National Cancer Institute (Cairo, Egypt). The viability of EAC cells was ensured employing Trypan blue dye exclusion method. The next step, EAC cells suspension was prepared in sterile saline solution to get a final working suspension; each 0.1 mL of which contained 2.5 million of EAC cells. At the beginning of the experiment, mice were shaved at their back, and inoculated with 0.1 mL of the EAC suspension.
Experimental DesignSeven days after inoculation with the tumor cells in all female mice, mice were randomly divided into fourteen groups, ten mice in each. The different treatments were started as follows; Group 1: mice treated daily with a 1 : 1 (v/v) mixture of DMSO/PEG400 (5 mL/kg/d, intraperitoneally (i.p.)). Group 2: mice treated daily with the standard chemotherapeutic agent, imatinib (10 mg/kg/d, i.p.). Groups 3–14: mice treated daily with compounds 2a–f, 4a, b, 5a, b, 6a and b (10 mg/kg/d, i.p.). All therapies continued for 10 d.
Dissection of Tumor Discs and Collection of Serum SamplesTumor MassOne day after the end of the experiment (day 18), mice were maintained under light ether anesthesia and blood samples were collected by cardiac puncture. Blood samples were maintained at room temperature for 30 min and centrifuged at 12000×g for 10 min. Then, sera were separated and stored at −20°C until used. An ELISA kit (Sun Red Biotechnology Company, Shanghai, China) was employed for estimating serum VEGF level according to the manufacturer’s protocol. The color intensity was measured at 450 nm using an automated ELISA reader (Europe S.A., Belgium).
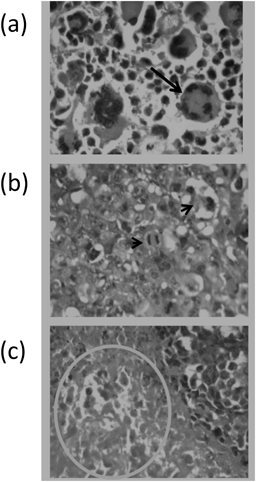
Histopathological Examination of the Solid TumorsHenceforward, mice were sacrificed by cervical dislocation under anesthesia and solid tumors from each mouse were dissected. Then, tumors were weighed and fixed in 10% phosphate-buffered formalin. All paraffin-embedded sections were cut at 4 µm and stained with hematoxylin and eosin (H&E). Histopathological evaluation for solid tumor sections was focused in the extension of necrotic area, finding of typical mitotic picture and the presence of neoplastic giant cells. Each one of these findings was scored according to their frequency and intensity: (0) absent, (1) weak or low, (2) mild to moderate and (3) high or frequent. Then, the summation of each score was calculated, averaged and compared for the experimental groups. Histopathological examinations were evaluated blindly.20)
Immunohistochemistry and Image AnalysisAnother tissue section (4 µm) was used for immunohistochemical staining of VEGF receptors-2. Briefly, rabbit polyclonal VEGF receptor-2 antibodies were added to each slide and incubated overnight at 4°C. Then, biotinylated secondary antibodies were added followed by the enzyme conjugate and finally, 2 drops of 3,3-diaminobenzidine chromogen were added to each slide to visualize the immune reaction. Finally, cover slipping was performed, the slides were examined blindly under a light microscope and photomicrographs were captured. The photomicrographs were then assessed using the ImageJ 1.45 F image analysis system (National Institute of Health, U.S.A.) to elucidate the percent of area of immunostaining.
Statistical AnalysisData were collected, tabulated and presented as the mean±standard error of the mean (S.E.M.) One-way ANOVA followed by Bonferroni’s post-hoc test were used to analyze the difference between the experimental groups. All statistical tests were performed employing the statistical package for social science (Chicago, IL, U.S.A.), version 16. A p value <0.05 was considered to be statistically significant.21)
VEGFR Inhibitory Activity Assay by ELISAVEGFR-3 tyrosine (Tyr) kinase inhibitory activity was determined using Abcam’s VEGF Receptor 3 Human ELISA (Sanfrancisco, CA, U.S.A.). The procedure of the used kit was performed according to the manufacturer’s instructions.
Molecular Modeling StudyDocking StepsAll the docking study was done using Molecular Operating Environment (MOE 2008.10: Chemical Computing Group, Canada) as the computational software. In order to perform docking, preliminary steps were done.
Enzyme structures were checked for missing atoms, bonds and contacts. All water of crystallization was deleted away from the active site except the one involved in the interaction with the ligand. Hydrogens and partial charges were added to the system, using Protonate 3D application. The active site was isolated; recognition of the amino acids and the backbone was hidden. The interactions of the ligand with the amino acids of the active site were studied.
Docking of CompoundsThe compounds were built using MOE molecular builder and energy minimized by Merck Molecular Force Field (MMFF94x). Hydrogens and partial charges were added to the system, using Protonate 3D application. Compounds were grouped in databases, and docked into the active site, using the MOE Dock tool.
Conformational Analysis of CompoundsThe algorithm generated conformations from a single 3D conformation by conducting a systemic search. In this way, all combinations of angles were created for each compound. A collection of poses was generated from the pool of ligand conformations using Triangle Matcher placement method. Poses were generated by superposition of ligand atom triplets and triplets of points in the receptor-binding site in a systemic way. Poses generated by placement methodology were scored using an available method implemented in MOE, the London dG scoring function, which estimates the free energy of binding of the ligand from a given pose. The top30 poses for each ligand were output in a MOE database. Each resulting pose was then subjected to MMFF94x energy minimization. The minimized docking conformations were then rescored, using London dG scoring method.




 OH/reflux 4 h, ii) ArNH2/CH3CH2CH2CH2OH/reflux 15h.
OH/reflux 4 h, ii) ArNH2/CH3CH2CH2CH2OH/reflux 15h.
 /C2H5OH/reflux 12 h, ii) RCOCl/ C2H5OH/reflux 9 h. iii) RNCS/C2H5OH/reflux 10 h.
/C2H5OH/reflux 12 h, ii) RCOCl/ C2H5OH/reflux 9 h. iii) RNCS/C2H5OH/reflux 10 h.