Abstract
Twelve guaiane-type sesquiterpenoids, including four new ones, 10-O-methyl-orientalol A (1), 10-O-ethyl-alismoxide (2), 3β,4β-expoxy-chrysothol (3), orientalol G (4), and a new norsesquiterpenoid, orientalol H (5), were isolated from Chinese Alismatis Rhizoma, the dried rhizome of Alisma orientale. The structures of new compounds were elucidated on the basis of spectroscopic data. Moreover, the inhibition of lipopolysaccharide (LPS)-induced nitric oxide (NO) production in RAW264.7 cells of all compounds was tested.
Alismatis Rhizoma (“Zexie” in Chinese), a Chinese crude drug prepared from dried rhizome of the aquatic plant Alisma orientale (SAM.) JUZEP. (Alismataceae) widely cultivated in China and Japan, has been used as a diuretic agent used to “remove dampness and promote water metabolism” in the body according to Chinese medicinal principles.1–5) Previous phytochemical and pharmacological study on Alismatis Rhizoma reported the isolation of prostane-type triterpenes and guaiane-type sesquiterpenes, which exhibited a series of bioactivities, such as diuretic, anti-hepatitis B virus (HBV), anti-bacterial, anti-hypertentive, hypolipidemic, anti-inflammatory, positive hypocholesterolemic activity, and repairing action to cholinergic acetyltransferase, etc.1–3,6–10) With the aim of discovering more structurally unique and bioactive terpenoids from natural resource, five new guaiane-type sesquiterpenes (1–5) along with seven known ones (6–12) (Fig. 1) were isolated from Alismatis Rhizoma. Furthermore, all compounds were examined for their anti-inflammatory potential and compounds 6 and 12 showed moderate inhibition of nitric oxide (NO) production with IC50 values of 36.997 and 48.602 µM, respectively. Herein, we report the isolation, structural elucidation and biological evaluation of the isolated compounds.
Results and Discussion
The powdered Alismatis Rhizoma was extracted with 95% EtOH, and the concentrated extract was suspended in H2O followed by partition with EtOAc. The EtOAc fraction was purified using various column chromatographies to yield twelve guaiane-type sesquiterpenoids (1–12).
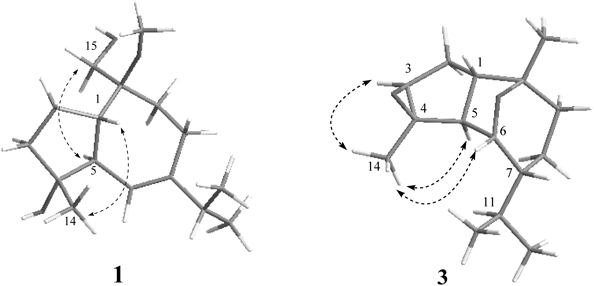
Compound 1, colorless oil, gave rise to a quasi-molecular ion peak at m/z 291.1935 ([M+Na]+) in the positive high resolution-electrospray ionization (HR-ESI)-MS, which corresponded to the molecular formula C16H28O3, in combination with its 13C-NMR and distortionless enhancement by polarization transfer (DEPT) analysis. The IR spectrum exhibited a broad absorption at 3420 cm−1 due to hydroxyl group. The 1H-NMR spectrum (Table 1) showed the presence of a tertiary methyl [δH 1.09 (3H, s)], a hydroxymethyl [δH 3.80 (1H, dd, 11.5, 6.8) and 3.75 (1H, dd, 11.5, 4.5)], two secondary methyls for an isopropyl group [δH 0.97 (3H, d, 6.8) and 0.98 (3H, d, 6.8)], an olefinic methine at δH 5.53 (br s) and a methoxyl group at δH 3.20 (3H, s). Besides the signals of the methoxyl group (δC 49.8, q), the 13C-NMR (DEPT) spectrum (Table 2) exhibited resonances for fifteen carbons: three methyls, one hydroxymethyl (δC 63.8), four methylenes, four methines including an olefinic one (δC 123.2), as well as two oxygenated quaternary carbons (δC 79.7 and 80.5) and an unsaturated quaternary carbon (δC 148.7), which were assigned to the guaiane-type sesquiterpenoid skeleton. The above NMR data of 1 were very similar to those of orientalol A (7),11) except for the appearance of the additional methoxyl group. Heteronuclear multiple bond connectivity (HMBC) correlations (Fig. 2) from H-6 (δH 5.53) to the signal at δC 79.7 (s), and from H-1 (δH 2.13), H-9 (δH 1.82 and 1.76) and H2-15 (δH 3.80 and 3.75) to the signal at δC 80.5 (s), unambiguously assigned the two oxygenated quaternary carbons as C-4 and C-10, respectively. The HMBC cross-peak of –OMe/C-10 revealed that the methoxyl group was attached to C-10, which was further supported by the variation of the chemical shift of C-1, C-9 and C-10. The other HMBC associated with correlated spectroscopy (COSY) (Fig. 2) permitted the completed assignment of the structure of 1. The relative stereochemistry of 1 was established by the coupling constants and the rotating frame nuclear Overhauser enhancement spectroscopy (ROESY) experiment (Fig. 3). The coupling constants of JH-1, H-5 (11.7 Hz) and JH-5, H-6 (0 Hz) revealed that the A/B ring junction was trans-oriented.12,13) The nuclear Overhauser effect (NOE) correlation from H-1, assumed to be β oriented, to Me-14, suggested that Me-14 was in β-configuration. The correlations from H-5 to H2-15 suggested that these protons were all in α-configuration. Thus, compound 1 was the 10-methoxyl derivative of compound 7, and finally concluded to be 4α,15-dihydroxy-10β-methoxy-1β,5α(H)-guai-6(7)-ene, named as 10-O-methyl-orientalol A.
Table 1.
1H-NMR Data of Compounds
1–
5 (δ in ppm,
J in Hz)
| No. | 1 (600 MHz, acetone-d6) | 2 (600 MHz, CDCl3) | 3 (800 MHz, acetone-d6) | 4 (600 MHz, acetone-d6) | 5 (600 MHz, acetone-d6) |
|---|
| 1 | 2.13 (1H, m) | 2.03 (1H, m) | 2.33 (1H, ddd, 11.0, 10.0, 5.1) | | 2.46 (1H, overlap) |
| 2 | 1.51 (1H, overlap) | 1.59 (1H, overlap) | 1.78 (1H, ddd, 14.9, 5.1, 1.5) | 2.49 (1H, dd, 7.5, 13.2) | 1.94 (1H, m) |
| 1.82 (1H, overlap) | 1.74 (1H, overlap) | 1.85 (1H, dd, 14.9, 10.0) | 1.76 (1H, m) | 1.79 (1H, overlap) |
| 3 | 1.51 (1H, overlap) | 1.60 (1H, overlap) | 3.54 (1H, d, 1.5) | 2.22 (1H, overlap) | 1.72 (1H, overlap) |
| 1.60 (1H, m) | 1.66 (1H, overlap) | | 2.30 (1H, overlap) | 1.82 (1H, overlap) |
| 5 | 2.42 (1H, d, 11.7) | 2.24 (1H, overlap) | 3.11 (1H, dd, 11.0, 7.9) | | 2.53 (1H, br d, 12.0) |
| 6 | 5.53 (1H, br s) | 5.46 (1H, d, 2.1) | 4.26 (1H, br d, 7.9) | 5.89 (1H, s) | 7.17 (1H, d, 3.0) |
| 7 | | | 1.25 (1H, m) | | |
| 8 | 1.95 (1H, dd, 16.6, 9.2) | 1.87 (1H, dd, 16.0, 9.8) | 1.63 (2H, m) | 4.27 (1H, d, 7.8) | 2.25 (1H, dd, 16.0, 9.8) |
| 2.23 (1H, overlap) | 2.23 (1H, overlap) | | | 2.68 (1H, dd, 16.0, 9.6) |
| 9 | 1.82 (1H, overlap) | 1.61 (1H, overlap) | 1.57 (2H, m) | 1.96 (1H, d, 12.5) | 2.07 (1H, m) |
| 1.76 (1H, m) | 1.72 (1H, overlap) | | 2.29 (1H, overlap) | 2.47 (1H, overlap) |
| 11 | 2.21 (1H, overlap) | 2.22 (1H, overlap) | 1.96 (1H, m) | 2.27 (1H, overlap) | |
| 12 | 0.97 (3H, d, 6.8) | 0.96 (3H, d, 6.5) | 0.98 (3H, d, 6.6) | 1.03 (3H, d, 6.4) | 2.31 (3H, s) |
| 13 | 0.98 (3H, d, 6.8) | 0.97 (3H, d, 6.5) | 0.96 (3H, d, 6.6) | 1.04 (3H, d, 6.4) | |
| 14 | 1.09 (3H, s) | 1.18 (3H, s) | 1.42 (3H, s) | 1.70 (3H, s) | 1.27 (3H, s) |
| 15 | 3.80 (1H, dd, 11.5, 6.8) | 1.20 (3H, s) | 1.11 (3H, s) | 1.09 (3H, s) | 4.75 (1H, s) |
| 3.75 (1H, dd, 11.5, 4.5) | | | | 4.80 (1H, s) |
| –OCH3 | 3.20 (3H, s) | | | | |
| –OCH2CH3 | | 3.36 (2H, m) | | | |
| –OCH2CH3 | | 1.13 (3H, t, 7.0) | | | |
| 4-OH | 3.52 (1H, br s) | | | | 3.87 (1H, br s) |
| 10-OH | | | | 3.58 (1H, br s) | |
Table 2.
13C-NMR Data of Compounds
1–
5 (δ in ppm)
| No. | 1 (150 MHz, acetone-d6) | 2 (150 MHz, CDCl3) | 3 (200 MHz, acetone-d6) | 4 (150 MHz, acetone-d6) | 5 (150 MHz, acetone-d6) |
|---|
| 1 | 47.0 (d) | 48.1 (d) | 53.3 (d) | 99.0 (s) | 46.6 (d) |
| 2 | 22.0 (t) | 21.7 (t) | 28.9 (t) | 28.3 (t) | 25.8 (t) |
| 3 | 41.1 (t) | 40.5 (t) | 69.6 (d) | 36.0 (t) | 40.4 (t) |
| 4 | 79.7 (s) | 80.3 (s) | 64.5 (s) | 135.4 (s) | 79.8 (s) |
| 5 | 50.5 (d) | 50.0 (d) | 56.9 (d) | 131.3 (s) | 56.9 (d) |
| 6 | 123.2 (d) | 121.1 (d) | 77.2 (d) | 111.8 (d) | 144.3 (d) |
| 7 | 148.7 (s) | 149.6 (s) | 41.7 (d) | 152.6 (s) | 144.9 (s) |
| 8 | 25.0 (t) | 24.7 (t) | 20.4 (t) | 74.7 (d) | 25.5 (t) |
| 9 | 31.7 (t) | 36.3 (t) | 31.0 (t) | 53.5 (t) | 36.7 (t) |
| 10 | 80.5 (s) | 79.0 (s) | 81.8 (s) | 80.3 (s) | 153.9 (s) |
| 11 | 38.0 (d) | 37.2 (d) | 28.6 (d) | 32.7 (d) | 198.5 (s) |
| 12 | 21.6 (q) | 21.2 (q) | 21.8 (q) | 21.4 (q) | 25.6 (q) |
| 13 | 22.0 (q) | 21.6 (q) | 21.6 (q) | 21.6 (q) | |
| 14 | 22.6 (q) | 22.4 (q) | 17.5 (q) | 13.6 (q) | 24.8 (q) |
| 15 | 63.8 (t) | 18.5 (q) | 27.7 (q) | 25.0 (q) | 107.5 (t) |
| –OCH3 | 49.8 (q) | | | | |
| –OCH2CH3 | | 55.9 (t) | | | |
| –OCH2CH3 | | 16.3 (q) | | | |
Compound 2 was assigned as a molecular formula of C17H30O2 by HR-ESI-MS which gave an ion peak [M+Na]+ at m/z 289.2154. The 1H- and 13C-NMR spectra (Tables 1, 2) showed signals of a guaiane sesquiterpene skeleton, which was close to 10-O-methyl-alismoxide,14) except the replacement of the methoxyl group with an ethoxyl group [δH 3.36 (2H, m), 1.13 (3H, t, 7.0); δC 55.9 (t), 16.3 (q)]. Two oxygenated quaternary carbons at δC 80.3 (s) and 79.0 (s) were assigned to be C-4 and C-10, respectively, according to the HMBC correlations of H-6/C-4, H-1/C-10, H-8/C-10, H-9/C-10 and Me-15/C-10. The ethoxyl group was located at C-10 based on the HMBC correlation from the protons at δH 3.36 to C-10. The coupling constants of JH-5, H-6 (2.1 Hz),12) together with a series of NOE correlations of H-1/Me-14, H-1/–OCH2CH3, H-1/–OCH2CH3, H-5/Me-15, indicated that compound 2 had the same relative configuration as that of 10-O-methyl-alismoxide. Consequently, compound 2 was identified as 4α-hydroxy-10β-ethoxy-1β,5α(H)-guai-6(7)-ene and named 10-O-ethyl-alismoxide.
The molecular formula C15H24O2 of 3 was determined by the HR-ESI-MS spectrum with an [M+Na]+ peak at m/z 259.1673, together with its 1H- and 13C-NMR data (Tables 1, 2), indicating four degrees of unsaturation. In the high field region of the 1H-NMR spectrum, two tertiary methyls (δH 1.42 and 1.11, each 3H, s) and two secondary methyls of the isopropyl group [δH 0.98 (3H, d, 6.6) and 0.96 (3H, d, 6.6)] was observed. 13C-NMR spectrum revealed that compound 3 contained fifteen carbons classified as four methyls, three methylenes, six methines (including two oxygenated ones at δC 69.6 and 77.2) and two oxygenated quaternary carbons at δC 64.5 and 81.8. The structure of 3 was thus tetracyclic due to the absence of any type of double bond. The presence of four oxygenic carbons and only two oxygen atoms in the molecular formula suggested the existence of two epoxy rings. Strong HMBC correlation (Fig. 2) between H-6 (δH 4.26) and C-10 (δC 81.8) revealed the ether linkage between C-6 and C-10. In addition, HMBCs from H-1, H-2 and H-5 to C-3 and C-4, from H-3 to C-1, C-2, C-14, and from Me-14 to C-3, C-4 and C-5, suggested that the other epoxy ring was located between C-3 and C-4. Furthermore, the structural units of C-3/C-2/C-1/C-5/C-6/C-7/C-8 and C-7/C-11/C-12 (C-13) were undoubtedly determined from the 1H–1H COSY spectrum (Fig. 2), which further supported the establishment of the structure. The coupling constant value of 11.0 Hz between H-1 and H-5 suggested that the two protons were trans-oriented.12,13) The NOE correlations (Fig. 3) from H-5, assumed to be α-oriented, to Me-14, and from H-3 and H-6 to Me-14, suggested that these protons and methyl were all in α-orientation. Consequently, two epoxy rings should be both in β-configuration. The coupling constant of H-6 and H-7 (J≈0 Hz) determined the β-orientation of H-7, thus, the isopropyl group at C-7 was located in α-orientation. Compound 3 was therefore elucidated to be 3β,4β-epoxy-6β,10β-epoxy-1β,5α,7β(H)-guaiane and named as 3β,4β-expoxy-chrysothol.
Compound 4 was isolated as colorless oil, whose molecular formula was determined to be C15H22O2 by a combination of HR-ESI-MS at m/z 257.1518 [M+Na]+ and NMR spectroscopic analysis, corresponding to five degrees of unsaturation. The 1H-NMR spectrum (Table 1) showed the presence of four methyls at δH 1.70 (3H, s), 1.09 (3H, s), 1.03 (3H, d, 6.4) and 1.04 (3H, d, 6.4), an olefinic methine at δH 5.89 (s) and an oxygenated methine at δH 4.27 (d, 7.8). The 13C-NMR (DEPT) spectrum (Table 2) revealed the presence of four methyls, three methenes, two methines including an oxygenated one (δC 74.7), two oxygenated quaternary carbons (δC 99.0 and 80.3), along with two double bonds [δC 111.8 (d), 131.3 (s), 135.4 (s) and 152.6 (s)]. The above spectral features suggested 4 to be a guaiane-type sesquiterpenoid, and the exact structure was determined by the following HMBC analysis (Fig. 2). Firstly, a series of HMBCs from H-6 to C-7, C-8 and C-11, from H-8 to C-6, C-7 and C-11, as well as from Me-12 and Me-13 to C-7 and C-11, indicated that one double bond was located between C-6 and C-7. Futhermore, the HMBC correlations of Me-14 with C-3, C-4 and C-5, and of H-6 with C-4 and C-5, suggested that the other double bond should exist between C-4 and C-5, and conjugated with the first one. Additionally, a broad singlet at 3.58 showed HMBC correlations with C-9, C-10 (δC 80.3) and Me-15, which assigned this signal as 10-OH. The correlations from Me-15 to C-9, C-10 and the other oxygenated quaternary carbons at δC 99.0 led to the assignment of this oxygenated quaternary carbons as C-1. Strong HMBC cross-peak between H-8 and C-1 illustrated the appearance of an oxygen bridge between C-1 and C-8, which was in accordance with the molecular formula of 4. The NOE correlation between H-8 and Me-15 suggested they were in the same side. Thus, the structure of 4 was finally determined as 10β-hydroxy-1β,8β-epoxy-guai-4(5),6(7)-diene, and named orientalol G.
Compound 5 had a molecular formula of C14H20O2, which was deduced from the pseudo-molecular ion m/z 243.1359 [M+Na]+ in its positive ion HR-ESI-MS spectrum, suggesting five degrees of unsaturation. The broad absorption at 3440 cm−1 and the strong absorption at 1667 cm−1 in its IR spectrum revealed the presence of hydroxyl and carbonyl groups, respectively. The 1H- and 13C-NMR spectral data (Tables 1, 2) of 5 showed the presence of two tertiary methyls, five methenes including an olefinic one [δH 4.75 (s) and 4.80 (s); δC 107.5 (t)], three methines containing an olefinic one [δH 7.17 (d, 3.0) and δC 144.3 (d)], as well as an oxygenated quaternary carbon (δC 79.8), two unsaturated quaternary carbons (δC 144.9 and 153.9), and a carbonyl group (δC 198.5). Extensive analysis of the above data suggested that compound 5 was a norsesquiterpenoid derivatived from alismol (6).10,11,14) The significant difference included the obvious presence of the carbonyl group and the lack of the isopropyl group. The HMBC correlations (Fig. 2) from Me-12 (δH 2.31), H-6 (δH 7.17) and H-8 [2.25 (dd, 9.8, 16.0) and 2.68 (dd, 9.6, 16.0)] to C-7 (δC 144.9) and the carbonyl group, suggested that the isopropyl group located at C-7 in 6 was replaced by the acetyl group [δC 198.5 (s, C-11) and 25.6 (q, C-12)]. The comparison of the NMR data of C-6, C-7, C-11 and C-12 in 5 with those of 4α,10β-dihydroxy-1βH,5αH-guai-6(7)-en-11-one, another norsesquiterpenoid isolated from Aglaia odorata var. microphyllina,13) further supported the existence of the α,β-unsaturated acetyl unit. The A/B ring junction was also trans-oriented based on the coupling constants of JH-1, H-5 (12.0 Hz).12,13) The NOE correlation of H-1 (β-oriented) with Me-14 suggested that Me-14 was also in β-orientation. Thus, the structure of 5 was undoubtedly established to be 4α-hydroxy-1β,5α(H)-guai-6(7),10(15)-dien-11-one, and named as orientalol H.
Unfortunately, the attempts to establish the absolute stereostructure of new compounds by single-crystal X-ray diffraction failed. In addition, since no any literature discussed the absolute configuration of guaiane-type sesquiterpenoids,1,11,13,15) the determination of the absolute stereochemistry of new compounds by calculated methods will be not very reliable. Thus, the absolute configuration of compounds 1–5 was not determined.
The known compounds were identified as alismol (6),10,11,14) orientalols A–C (7–9),11) orientalol E (10),10) alismoxide (11)10,11,14,16) and 4-epi-alismoxide (12)16) by comparison of their spectral data with literature values.
Compounds 1–12 were tested for inhibition of NO production in lipopolysaccharide (LPS)-stimulated RAW264.7 cells to evaluate their anti-inflammatory activity. Among the compounds tested, compounds 6 and 12 exhibited moderate inhibition of NO production with IC50 values of 36.997 and 48.602 µM, respectively, while the IC50 value of the positive control (NG-monomethyl-L-arginine, monoacetate salt (L-NMMA)) was 11.318 µM.
Experimental
General Experimental ProceduresOptical rotations were recorded on a Jasco P-1020 polarimeter (JASCO Corporation, Tokyo, Japan). UV spectra were measured using a Shimadzu UV-2401A spectrophotometer (Shimadzu Corporation, Kyoto, Japan). IR spectra were acquired using Bio-Rad FTS-135 spectrophotometer with KBr pellets (Bio-Rad Corporation, CA, U.S.A). NMR spectra were performed on Bruker AV 600 and 800 MHz spectrometers with tetramethylsilane (TMS) as an internal standard (Bruker BioSpin Group, Germany). ESI-MS and HR-ESI-MS were obtained on an API-Qstar-Pulsar spectrometer (Applied Biosystems Corporation, Canada). Sephadex LH-20 (Amersham Biosciences AB, Uppsala, Sweden), silica gel (200–300 mesh, Qingdao Marine Chemical Co., Ltd., P. R. China), ODS-C18 (75 µm, YMC Co., Ltd., Japan) and MCI gel (CHP 20P, 75–150 µm, Mitsubishi Chemical Corporation, Tokyo, Japan) were used for column chromatography (CC). Semi-preparative HPLC was carried out on an Agilent 1200 LC with a Zorbax SB-C18 (5 µm, 9.4×250 mm, Agilent, U.S.A.) column. Fractions were monitored by TLC plates (Si gel GF254, Qingdao Marine Chemical Co., Ltd., P. R. China), and spots were visualized by heating silica gel plates sprayed with 5% H2SO4–EtOH.
Plant MaterialAlismatis Rhizoma were purchased from a market of Chinese medical materials located at Luosiwan International Trade City, Kunming, P. R. China, in September 2015, and identified by Prof. Xiao Cheng, Kunming Institute of Botany, Chinese Academy of Sciences. A voucher specimen (KIB20150901) has been deposited at the State Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany, Chinese Academy of Sciences.
Extraction and IsolationThe powdered Alismatis Rhizoma (10 kg) were extracted with 95% aq. EtOH (3×10 L, 24 h, each) at room temperature (r.t.), and then concentrated under vacuum to yield a crude extract, which was suspended in H2O followed by partition with EtOAc. The EtOAc extract (398 g) was subjected to medium-pressure liquid chromatography (MPLC) over MCI gel (60, 70, 80, 90 and 100% MeOH/H2O, 16 L for each gradient) to afford six fractions, Fr. A–F. The purification of Fr. A (60% MeOH/H2O, 18 g) by Sephadex LH-20 (MeOH), silica gel CC (CHCl3–MeOH 20 : 1 and CHCl3–isopropanol 20 : 1) and semi-preparative HPLC (45% MeOH/H2O) led to the isolation of compound 8 (retention time (tR)=28.0 min, 6.0 mg). Fr. B-1–B-5 were obtained from Fr. B (70% MeOH/H2O, 8.7 g) by silica gel CC (CHCl3–acetone 20 : 1). Through repeated silica gel CC eluting with petroleum ether–acetone 15 : 1 and CHCl3–isopropanol 100 : 1, compound 10 (7.0 mg) was obtained from Fr. B-3 (285 mg). Fr. C (70% MeOH/H2O, 58 g) was divided into twelve parts (Fr. C-1–C-12) by MPLC octadecylsilyl (ODS) eluted with gradient MeOH/H2O (40, 45, 50, 55, 60, 65, 70, 75, 80%, 4L for each gradient). Fr. C-1 (1.2 g) was separated by silica gel CC (CHCl3–MeOH 50 : 1) and then purified by semi-preparative HPLC (50% MeOH/H2O) to obtain compound 7 (tR=12.0 min, 17.0 mg). Fr. C-2 (0.5 g) was chromatographed over repeated silica gel CC (petroleum ether–acetone 7 : 1 and CHCl3–MeOH 80 : 1) and followed by Sephadex LH-20 using MeOH as elution to produce compound 11 (2.0 mg). Fr. C-3 (1.6 g) was separated into Fr. C-3-1 (108 mg) and Fr. C-3-2 (36 mg) by silica gel CC (petroleum ether–acetone 9 : 1). Both subfractions were purified over semi-preparative HPLC (30% MeCN/H2O and 53% MeOH/H2O, respectively) to give compounds 5 (tR=24.0 min, 4.4 mg) and 1 (tR=25.4 min, 6.0 mg). Fr. C-5 (5.5 g) was submitted to silica gel CC (petroleum ether–acetone 12 : 1) to get five fractions, Fr. C-5-1–C-5-5. Fr. C-5-1 (220 mg) was further fractionated by silica gel CC using petroleum ether–acetone 60 : 1 and pure CHCl3 successively to afford compound 3 (1.6 mg). Compound 4 (tR=24.0 min, 5.7 mg) was obtained from Fr. C-5-2 (278 mg) via silica gel CC (petroleum ether–CHCl3–isopropanol 50 : 50 : 1) and semi-preparative HPLC (60% MeOH/H2O). Fr. C-6 (12.5 g) was separated to eleven parts (Fr. C-6-1–C-6-11) by silica gel CC (petroleum ether–acetone 7 : 1). Compound 12 (tR=31.3 min, 4.7 mg) was produced from Fr. C-6-1 (105 mg) on semi-preparative HPLC (60% MeOH/H2O). Fr. C-6-2 (609 mg) was successively applied to silica gel CC (petroleum ether–acetone 15 : 1), Sephadex LH-20 (MeOH) and semi-preparative HPLC (38% MeCN/H2O) to get compound 9 (tR=27.3 min, 12.0 mg). Taken the same method as Fr. C-6, Fr. C-7 (5.5 g) was subjected to silica gel CC (petroleum ether–acetone 7 : 1) to give eight fractions, Fr. C-7-1–C-7-8. Through silica gel (petroleum ether–acetone 40 : 1) and semi-preparative HPLC (70% MeOH/H2O), Fr. C-7-1 (385 mg) gave compound 2 (tR=23.6 min, 22.0 mg). Fr. D (80% MeOH/H2O, 79 g) was submitted to silica gel CC eluting with CHCl3–isopropanol (gradient 100 : 1→1 : 1) to obtain subfractions D-1–D-10. Fr. D-2 (15 g) was further fractionated by silica gel CC (petroleum ether–acetone, 20 : 1, 9 : 1, 8 : 2) to yield compound 6 (1.7 g).
10-O-Methyl-orientalol A (1)Colorless oil; [α]D26 2.35 (c=0.14, MeOH). IR (KBr) cm−1: 3420, 3054, 2959, 1708, 1628, 1463, 1382, 1077, 957. UV λmax (MeOH) nm (log ε): 204.0 (3.69). 1H-NMR (600 MHz, acetone-d6) and 13C-NMR (150 MHz, acetone-d6) spectroscopic data, see Tables 1 and 2. HR-ESI-MS (pos.) m/z: 291.1935 [M+Na]+ (Calcd for C16H28O3Na: 291.1931).
10-O-Ethyl-alismoxide (2)Colorless oil; [α]D26−0.24 (c=0.25, MeOH). IR (KBr) cm−1: 3417, 2966, 2870, 1630, 1462, 1384, 1301, 1103, 1090, 1067, 956. UV λmax (MeOH) nm (log ε): 203.4 (3.25). 1H-NMR (600 MHz, CDCl3) and 13C-NMR (150 MHz, CDCl3) spectroscopic data, see Tables 1 and 2. HR-ESI-MS (pos.) m/z: 289.2154 [M+Na]+ (Calcd for C17H30O2Na: 289.2138).
3β,4β-Expoxy-chrysothol (3)Colorless oil; [α]D26−2.74 (c=0.09, MeOH). 1H-NMR (800 MHz, acetone-d6) and 13C-NMR (200 MHz, acetone-d6) spectroscopic data, see Tables 1 and 2. HR-ESI-MS (pos.) m/z: 259.1673 [M+Na]+ (Calcd for C15H24O2Na: 259.1669).
Orientalol G (4)Colorless oil; [α]D26−7.67 (c=0.16, MeOH). UV λmax (MeOH) nm (log ε): 203.0 (3.62), 232.0 (sh), 282.5 (sh). 1H-NMR (600 MHz, acetone-d6) and 13C-NMR (150 MHz, acetone-d6) spectroscopic data, see Tables 1 and 2. HR-ESI-MS (pos.) m/z: 257.1518 [M+Na]+ (Calcd for C15H22O2Na: 257.1512).
Orientalol H (5)Colorless oil; [α]D26−4.23 (c=0.09, MeOH). IR (KBr) cm−1: 3440, 3082, 2964, 2933, 2872, 1667, 1441, 1384, 1267, 1213, 1126, 933, 890. UV λmax (MeOH) nm (log ε): 201.0 (3.98), 234.2 (4.16). 1H-NMR (600 MHz, acetone-d6) and 13C-NMR (150 MHz, acetone-d6) spectroscopic data, see Tables 1 and 2. HR-ESI-MS (pos.): 243.1359 [M+Na]+ (Calcd for C14H20O2Na: 243.1356).
Inhibition of Nitric Oxide Production AssayMurine macrophage RAW264.7 cells (Kunming Institute of Zoology, Chinese Academy of Sciences) were maintained in Dulbecco’s modified Eagle’s medium (DMEM, high glucose) supplemented with 10% (v/v) fetal bovine serum, 1% penicillin (10000 U/mL)–streptomycin (10000 µg/mL), along with 10 mmol/L N-(2-hydroxyethyl)-piperazine-N′-2-ethanesulfonic acid (HEPES) in a 37°C, 5% CO2 incubator. Inhibition of NO production assay was performed as described previously.17) Cells were pre-treated with compounds 1–12 at the concentrations of 0, 0.39, 1.56, 6.25, 25 and 100 µM, and co-incubated with 1 µg/mL LPS for 24 h. Dimethyl sulfoxide (DMSO) and L-NMMA were used as negative and positive control, respectively. Cytotoxicity was determined by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay as described.18)
Acknowledgments
This work was financially supported by the National Natural Science Foundation of China (Nos. U1502223 and 31560103) and the National Basic Research Program of China (973 Program No. 2011CB915503).
Conflict of Interest
The authors declare no conflict of interest.
Supplementary Materials
The online version of this article contains supplementary materials.
References
- 1) Jiang Z. Y., Zhang X. M., Zhou J., Zhang F. X., Chen J. J., Lü Y., Wu L., Zheng Q. T., Chem. Pharm. Bull., 55, 905–907 (2007).
- 2) Fukuyama Y., Geng P. W., Wang R., Yamada T., Nakagawa K., Planta Med., 54, 445–447 (1988).
- 3) Zhao M., Xu L. J., Che C. T., Phytochemistry, 69, 527–532 (2008).
- 4) Yoshikawa M., Hatakeyama S., Tanaka N., Fukuda Y., Yamahara J., Murakami N., Chem. Pharm. Bull., 41, 1948–1954 (1993).
- 5) Chinese Pharmacopoeia Commission, “Pharmacopoeia of the People’s Republic of China,” Vol. 1, China Medical Science Press, Beijing, 2015, p. 229.
- 6) Lee S. M., Kho Y. H., Min B. S., Kim J. H., Na M. K., Kang S. J., Maeng H. Y., Bae K. H., Arch. Pharm. Res., 24, 524–526 (2001).
- 7) Jin H. G., Jin Q., Ryun Kim A., Choi H., Lee J. H., Kim Y. S., Lee D. G., Woo E. R., Arch. Pharm. Res., 35, 1919–1926 (2012).
- 8) Yoshikawa M., Hatakeyama S., Tanaka N., Matsuoka T., Yamahara J., Murakami N., Chem. Pharm. Bull., 41, 2109–2112 (1993).
- 9) Yoshikawa M., Tomohiro N., Murakami T., Ikebata A., Matsuda H., Matsuda H., Kubo M., Chem. Pharm. Bull., 47, 524–528 (1999).
- 10) Peng G. P., Lou F. C., Nat. Prod. Res. Dev., 13, 1–3 (2001).
- 11) Yoshikawa M., Hatakeyama S., Tanaka N., Fukuda Y., Murakami N., Yamahara J., Chem. Pharm. Bull., 40, 2582–2584 (1992).
- 12) Anjaneyulu A. S. R., Gowri P. M., Indian J. Chem., 39B, 773–778 (2000).
- 13) Liu S., Liu S. B., Zuo W. J., Guo Z. K., Mei W. L., Dai H. F., Fitoterapia, 92, 93–99 (2014).
- 14) Nakajima Y., Satoh Y., Katsumata M., Tsujiyama K., Ida Y., Shoji J., Phytochemistry, 36, 119–127 (1994).
- 15) Ma Q. J., Han L., Bi X. X., Wang X. B., Mu Y., Guan P. P., Li L. Y., Huang X. S., Phytochemistry, 131, 150–157 (2016).
- 16) Blay G., García B., Molina E., Pedro J. R., J. Org. Chem., 71, 7866–7869 (2006).
- 17) Huang G. J., Pan C. H., Liu F. C., Wu T. S., Wu C. H., Food Chem. Toxicol., 50, 1485–1493 (2012).
- 18) Israf D. A., Khaizurin T. A., Syahida A., Lajis N. H., Khozirah S., Mol. Immunol., 44, 673–679 (2007).