Experimental
General RemarksMelting points (mp) were measured with a Yanaco MP-J3 melting point apparatus and are uncorrected. NMR spectra were recorded on a Bruker AVANCE500 (500 MHz for 1H and 126 MHz for 13C) with tetramethylsilane (Me4Si) as an internal reference and CDCl3 as the solvent. 1H- and 13C-NMR spectral data are reported in parts per million (δ) relative to Me4Si. IR spectra were recorded on a JASCO FT/IR-4100 spectrometer. Mass spectra were recorded on JEOL JMS-GC mate II spectrometer with a direct inlet system at 70 eV and a Bruker micrOTOF-Q mass spectrometer with methanol as the solvent. Elemental analyses were carried out on a Yanaco CHN Corder MT-5 at the Integrated Center for Science, Ehime University. Standard work-up means that the organic layers were finally dried over anhyd. Na2SO4, filtered, and concentrated in vacuo below 37°C using a rotary evaporator.
MaterialsThe following compounds were prepared by employing the reported method.
N-Benzoyl-N-methylglycine (2a)mp 101–104°C (mp23) 102–104°C).
N-Benzoyl-N-phenylglycine (2b)mp 126–128°C (mp23) 127–129°C).
N-Acetyl-N-phenylglycine (2c)mp 196–198°C (mp24) 193–195°C).
N-(4-Bromobenzoyl)-N-methylglycine (2d)mp 143–145°C (mp25) 147–150°C).
N-(4-Methoxybenzoyl)-N-methylglycine (2e)mp 151–154°C (mp23) 155–160°C).
N-Benzoyl-N-benzylglycine (2f)mp 103–104°C (mp26) 106–107°C).
N-Benzoyl-N-(4-methoxyphenylmethyl)glycine (2g)mp 157–158°C (mp27) 157–158°C).
N-Benzyl-N-(4-methoxybenzoyl)glycine (2h)mp 149–151°C (mp25) 149–151°C).
N-Methyl-N-pivaloylglycine (2i)mp 65–68°C (mp28) 75–76°C).
N-(4-Nitrobenzoyl)-N-phenylglycine (2j)mp 178–180°C (mp23) 172–175°C).
N-(4-Methoxybenzoyl)-N-phenylglycine (2k)mp 135–137°C (mp23) 158–163°C).
4-Trifluoroacetyl-3-methyl-2-phenyl-1,3-oxazolium-5-olate (1a)Pale yellow crystals, 95% yield. mp 161–163°C (mp29) 162–163°C).
4-Trifluoroacetyl-2,3-diphenyl-1,3-oxazolium-5-olate (1b)Yellow crystals, 94% yield. mp 194–196°C (mp29) 194–196°C).
4-Trifluoroacetyl-2-methyl-3-phenyl-1,3-oxazolium-5-olate (1c)White crystals, 90% yield. mp 200–203°C (mp29) 211–212°C).
2-(4-Bromophenyl)-4-trifluoroacetyl-3-methyl-1,3-oxazolium-5-olate (1d)White crystals, 90% yield. mp 188–191°C (mp25) 188–191°C).
4-Trifluoroacetyl-2-(4-methoxyphenyl)-3-methyl-1,3-oxazolium-5-olate (1e)Pale yellow crystals, 82% yield. mp 142–143°C (ethyl acetate/hexane). (mp17) 142–143°C).
3-Benzyl-4-trifluoroacetyl-2-phenyl-1,3-oxazolium-5-olate (1f)White crystals, 83% yield. mp 143–145°C (mp27) 143–145°C).
4-Trifluoroacetyl-3-(4-methoxybenyl)-2-phenyl-1,3-oxazolium-5-olate (1g)White crystals, 75% yield. mp 147–148°C (mp27) 147–148°C).
3-Benzyl-4-trifluoroacetyl-2-(4-methoxyphenyl)-1,3-oxazolium-5-olate (1h)White crystals, 89% yield. mp 158–160°C (mp26) 158–160°C).
2-tert-Butyl-4-trifluoroacetyl-3-methyl-1,3-oxazolium-5-olate (1i)White crystals, 67% yield. mp 120–121°C (mp28) 120–121°C).
4-Trifluoroacetyl-2-(4-nitrophenyl)-3-phenyl-1,3-oxazolium-5-olate (1j)Yellow crystals, 70% yield. mp 175–178°C (ethyl acetate/hexane). (mp17) 175–178°C).
4-Trifluoroacetyl-2-(4-methoxyphenyl)-3-phenyl-1,3-oxazolium-5-olate (1k)Pale yellow crystals, 93% yield. mp 203–205°C (mp25) 182–185°C).
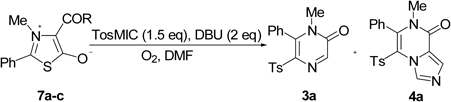
General Procedure for Synthesis of 2(1H)-Pyrazinones (3) and Imidazo[1,5-a]pyrazin-8(7H)-ones (4)To a stirred solution of TosMIC (146 mg, 0.75 mmol) in DMF (3 mL) was added DBU (152 mg, 1.00 mmol) at 0°C, and the mixture was stirred for 1 h under atmosphere of oxygen. To the mixture was added 4-trifluoroacetyl-1,3-oxazolium-5-olate 1 (0.50 mmol), and the whole was stirred at 0°C for an additional several hours. After workup with aq. Na2CO3, the mixture was extracted with AcOEt (×3). The combined organic layers were washed with brine, dried over anhyd. Na2SO4, and evaporated. The residue was purified by column chromatography (silica gel, hexane–AcOEt=4 : 1 to 1 : 1) to give the products 3 and 4.
1-Methyl-5-[(4-methylphenyl)sulfonyl]-6-phenylpyrazin-2(1H)-one (3a)White crystals, 72% yield. mp 147–149°C (CHCl3–hexane). IR (KBr) cm−1: 3057, 1674, 1662, 1487, 1322, 575. 1H-NMR (500 MHz, CDCl3) δ: 2.41 (s, 3H, ArCH3), 3.13 (s, 3H, NCH3), 7.21–7.27 (m, 4H, ArH), 7.51–7.59 (m, 5H, ArH), 8.13 (s, 1H, H-3). 13C-NMR (126 MHz, CDCl3) δ: 21.5 (CH3), 33.3 (NCH3), 128.3 (CH), 128.6 (CH), 128.9 (CH), 129.0 (C), 129.4 (CH), 130.6 (CH), 134.0 (C-5), 137.3 (C), 144.3 (C), 145.0 (C-6), 145.8 (C-3), 155.5 (CO). MS m/z: 313 (M+, 66), 58 (100). Anal. Calcd for C18H16N2O3S: C, 63.51; H, 4.74; N, 8.23. Found: C, 63.32; H, 5.03; N, 8.09.
5-[(4-Methylphenyl)sulfonyl]-1,6-diphenylpyrazin-2(1H)-one (3b)Yellow crystals, 65% yield. mp 228–230°C (CHCl3–hexane). IR (KBr) cm−1: 3059, 3050, 1685, 1485, 1328, 1214, 1169, 1160, 806, 692, 681, 651, 585, 553. 1H-NMR (500 MHz, CDCl3) δ: 2.43 (s, 3H, ArCH3), 6.89–6.91 (m, 2H, ArH), 7.02–7.04 (m, 2H, ArH), 7.17–7.27 (m, 8H, ArH), 7.62 (d, J=8.0 Hz, 2H, ArH), 8.24 (s, 1H, H-3). 13C-NMR (126 MHz, CDCl3) δ: 21.7 (CH3), 127.7 (CH), 128.3 (CH), 128.6 (CH), 128.7 (C), 129.2 (C), 129.2 (CH), 129.5 (CH), 129.8 (CH), 130.0 (CH), 134.0 (C), 135.3 (C), 137.3 (C), 144.5 (CH), 144.8 (C), 147.6 (C-3), 155.2 (CO). MS m/z: 402 (M+, 5.6), 180 (100). Anal. Calcd for C23H18N2O3S·0.5H2O: C, 67.14; H, 4.65; N, 6.81. Found: C, 66.80; H, 4.73; N, 6.75.
6-Methyl-5-[(4-methylphenyl)sulfonyl]-1-phenylpyrazin-2(1H)-one (3c)Orange crystals, 42% yield. mp 212–215°C (CHCl3–hexane). IR (KBr) cm−1: 2926, 1680, 1508, 1491, 1305, 1159, 696, 675, 586, 556. 1H-NMR (500 MHz, CDCl3) δ: 2.46 (s, 6H, CH3 and ArCH3), 7.13 (d, J=7.5 Hz, 2H, ArH), 7.37 (d, J=8.0 Hz, 2H, ArH), 7.52–7.59 (m, 3H, ArH), 7.91 (d, J=8.5 Hz, 2H, ArH), 8.03 (s, 1H, H-3). 13C-NMR (126 MHz, CDCl3) δ: 17.3 (CH3), 21.7 (CH3), 127.2 (CH), 128.5 (CH), 129.8 (CH), 130.2 (C), 130.5 (CH), 133.7 (C), 135.5 (C), 137.1 (C), 143.6 (C), 144.8 (C), 145.4 (C-3), 155.5 (CO). MS m/z: 340 (M+, 11.7), 118 (100). Anal. Calcd for C18H16N2O3S·0.5H2O: C, 61.87; H, 4.90; N, 8.02. Found: C, 62.21; H, 4.61; N, 8.24.
6-(4-Bromophenyl)-1-methyl-5-[(4-methylphenyl)sulfonyl]pyrazin-2(1H)-one (3d)Pale yellow crystals, 70% yield. mp 178–180°C (CHCl3–hexane). IR (KBr) cm−1: 3050, 2953, 2922, 1676, 1593, 1483, 1323, 1136, 1010, 850, 655, 582. 1H-NMR (500 MHz, CDCl3) δ: 2.42 (s, 3H, ArCH3), 3.14 (s, 3H, NCH3), 7.13 (dd, J=8.4, 1.9 Hz, 2H, ArH), 7.26–7.27 (m, 2H, ArH), 7.60 (dd, J=8.3, 1.7 Hz, 2H ArH), 7.69 (dd, J=8.5, 1.9 Hz, 2H, ArH), 8.13 (s, 1H, H-3). 13C-NMR (126 MHz, CDCl3) δ: 21.7 (CH3), 33.4 (NCH3), 125.3 (C), 127.9 (C), 128.3 (CH), 129.6 (CH), 130.3 (CH), 132.3 (CH), 134.0 (C), 137.1 (C), 143.8 (C), 144.7 (C), 146.2 (C-3), 155.4 (CO). MS m/z: 418 (4.2)+420 (4.3) (M+, 1 : 1), 196 (100). Anal. Calcd for C18H15BrN2O3S·0.5H2O: C, 50.48; H, 3.77; N, 6.54. Found: C, 50.47; H, 3.77; N, 6.68.
6-(4-Methoxyphenyl)-1-methyl-5-[(4-methylphenyl)sulfonyl]pyrazin-2(1H)-one (3e)Pale yellow crystals, 78% yield. mp 135–138°C (CHCl3–hexane). IR (KBr) cm−1: 2955, 2833, 1671, 1497, 1257, 1178, 1152, 848, 658. 1H-NMR (500 MHz, CDCl3) δ: 2.40 (s, 3H, ArCH3), 3.15 (s, 3H, NCH3), 3.90 (s, 3H, OCH3), 7.02 (d, J=8.7 Hz, 2H, ArH), 7.13 (d, J=8.6 Hz, 2H, ArH), 7.23 (d, J=8.2 Hz, 2H, ArH), 7.57 (d, J=8.2 Hz, 2H, ArH), 8.11 (s, 1H, H-3). 13C-NMR (126 MHz, CDCl3) δ: 21.6 (CH3), 33.3 (NCH3), 55.4 (OCH3), 114.3 (CH), 120.8 (C), 128.2 (CH), 129.4 (CH), 130.2 (CH), 134.4 (C), 137.5 (C), 144.3 (C), 145.3 (C), 145.5 (C-3), 155.8 (CO), 161.2 (C). MS m/z: 370 (M+, 5.1), 148 (100). Anal. Calcd for C19H18N2O4S·0.5H2O: C, 60.14; H, 5.05; N, 7.38. Found: C, 60.02; H, 5.06; N, 7.35.
1-Benzyl-5-[(4-methylphenyl)sulfonyl]-6-phenylpyrazin-2(1H)-one (3f)White crystals, 55% yield. mp 164–165°C (CHCl3–hexane). IR (KBr) cm−1: 3063, 3046, 3030, 1668, 1327, 1162, 704, 654, 576. 1H-NMR (500 MHz, CDCl3) δ: 2.41 (s, 3H, ArCH3), 4.95 (s, 2H, NCH2Ar), 6.75 (d, J=7.0 Hz, 2H, ArH), 7.00 (d, J=7.0 Hz, 2H, ArH), 7.05–7.30 (m, 5H, ArH), 7.37 (t, J=8.0 Hz, 2H, ArH), 7.54 (m, 3H, ArH), 8.21 (s, 1H, H-3). 13C-NMR (126 MHz, CDCl3) δ: 21.6 (CH3), 48.7 (NCH2), 127.3 (CH), 128.0 (CH), 128.1 (C), 128.3 (CH), 128.4 (CH), 128.5 (CH), 129.4 (CH), 129.5 (CH), 130.6 (CH), 134.4 (C), 134.7 (C), 137.3 (C), 144.5 (C), 145.0 (C), 146.8 (C-3), 155.6 (CO). MS m/z: 416 (M+, 22.8), 91 (100). Anal. Calcd for C24H20N2O3S: C, 69.21; H, 4.84; N, 6.73. Found: C, 69.21; H, 4.79; N, 6.86.
1-(4-Methoxyphenylmethyl)-5-[(4-methylphenyl)sulfonyl]-6-phenylpyrazin-2(1H)-one (3g)Pale yellow oil, 53% yield. IR (neat) cm−1: 3062, 2957, 3014, 1674, 1514, 1327, 1250, 1162, 795. 1H-NMR (500 MHz, CDCl3) δ: 2.40 (s, 3H, ArCH3), 3.75 (s, 3H, NCH3), 4.89 (s, 2H, NCH2Ar), 6.69 (s, 4H, ArH), 7.03 (d, J=8.3 Hz, 2H, ArH), 7.22 (d, J=8.1 Hz, 2H, ArH), 7.41 (t, J=8.1 Hz, 2H, ArH), 7.53–7.56 (m, 3H, ArH), 8.19 (s, 1H, H-3). 13C-NMR (126 MHz, CDCl3) δ: 21.6 (CH3), 48.3 (NCH2), 55.3 (OCH3), 113.8 (CH), 126.7 (C), 128.1 (C), 128.3 (CH), 128.4 (CH), 129.1 (CH), 129.5 (CH), 129.5 (CH), 130.6 (C), 134.3 (C), 137.3 (C), 144.5 (CH), 145.0 (C), 146.7 (C-3), 155.7 (CO), 159.3 (C). MS m/z: 446 (M+, 15.4), 121 (100). HR-MS (EI) for C25H22N2O4S (M+): Calcd, 446.1300. Found, 446.1307.
1-Benzyl-6-(4-methoxyphenyl)-5-[(4-methylphenyl)sulfonyl]pyrazin-2(1H)-one (3h)Yellow oil, 58% yield. IR (neat) cm−1: 3063, 3030, 2964, 1695, 1681, 1651. 1H-NMR (500 MHz, CDCl3) δ: 2.40 (s, 3H, ArCH3), 3.89 (s, 3H, ArOCH3), 4.96 (s, 2H, NCH2Ar), 6.79 (d, J=8.2 Hz, 2H, ArH), 6.85–6.93 (m, 4H, ArH), 7.17–7.26 (m, 5H, ArH), 7.55 (d, J=8.3 Hz, 2H, ArH), 8.19 (s, 1H, ArH). 13C-NMR (126 MHz, CDCl3) δ: 21.6 (CH3), 48.7 (NCH2), 55.4 (OCH3), 113.7 (CH), 120.1 (C), 127.3 (CH), 128.0 (C), 128.4 (CH), 128.6 (CH), 129.5 (CH), 130.9 (CH), 134.8 (CH), 134.9 (C), 137.3 (C), 144.4 (C), 145.2 (C), 146.5 (C-3), 155.8 (CO), 161.2 (C). MS m/z: 446 (M+, 40.4), 91 (100). HR-MS (EI) for C25H22N2O4S (M+): Calcd, 446.1281. Found, 446.1300.
6-tert-Butyl-1-methyl-5-[(4-methylphenyl)sulfonyl]pyrazin-2(1H)-one (3i)Pale yellow crystals, 11% yield. mp 128–130°C (CHCl3–hexane). IR (KBr) cm−1: 2981, 1668, 1482, 1301, 1162, 658. 1H-NMR (500 MHz, CDCl3) δ: 1.80 (s, 9H, C(CH3)3), 2.43 (s, 3H, ArCH3), 3.64 (s, 3H, NCH3), 7.29 (d, J=8.0 Hz, 2H, ArH), 7.62 (s, 1H, H-3), 7.77 (d, J=8.0 Hz, 2H, ArH). 13C-NMR (126 MHz, CDCl3) δ: 21.6 (CH3), 31.3 (C(CH3)), 38.3 (C(CH3)), 39.6 (NCH3), 129.2 (CH), 129.2 (CH), 138.0 (C), 139.0 (C), 139.8 (CH), 144.2 (C), 156.6 (C), 156.8 (C). MS m/z: 320 (M+, 1.6), 240 (100). Anal. Calcd for C16H20N2O3S·0.5H2O: C, 58.34; H, 6.43; N, 8.50. Found: C, 58.03; H, 6.14; N, 8.32.
5-[(4-Methylphenyl)sulfonyl]-6-(4-nitrophenyl)-1-phenylpyrazin-2(1H)-one (3j)Pale orange crystals, 25% yield. mp >300°C (dec.) (CHCl3–hexane). IR (KBr) cm−1: 3065, 1693, 1520, 1489, 1350, 1329, 1166, 862, 701. 1H-NMR (500 MHz, CDCl3) δ: 2.45 (s, 3H, ArCH3), 6.93 (dd, J=8.0, 2.2 Hz, 2H, ArH), 7.25–7.34 (m, 7H, ArH), 7.69 (d, J=8.3 Hz, 2H ArH), 8.09 (d, J=8.7 Hz, 2H, ArH), 8.25 (s, 1H, H-3). 13C-NMR (126 MHz, CDCl3) δ: 21.7 (CH3), 122.9 (CH), 128.1 (CH), 128.7 (CH), 129.7 (CH), 129.8 (CH), 129.9 (C), 131.1 (CH), 133.8 (C), 134.7 (C), 135.2 (C), 136.6 (C), 142.1 (C), 145.2 (CH), 148.2 (C), 148.7 (C-3), 154.6 (CO). MS m/z: 447 (M+, 1.5), 77 (100). Anal. Calcd for C23H17N3O5S·0.33H2O: C, 61.10; H, 4.33; N, 9.32. Found: C, 61.14; H, 4.22; N, 9.26.
6-(4-Methoxyphenyl)-5-[(4-methylphenyl)sulfonyl]-1-phenylpyrazin-2(1H)-one (3k)Pale orange crystals, 35% yield. mp 202–206°C (dec.) (CHCl3–hexane). IR (KBr) cm−1: 3026, 1676, 1596, 1446, 1359, 1340, 1258, 1162, 1087, 885, 810, 789, 658. 1H-NMR (500 MHz, CDCl3) δ: 2.42 (s, 3H, ArCH3), 3.75 (s, 3H, OCH3), 6.69 (d, J=8.7 Hz, 2H, ArH), 6.90 (dd, J=8.3, 1.7 Hz, 2H, ArH), 6.93 (d, J=8.7 Hz, 2H, ArH), 7.22–7.27 (m, 5H, ArH), 7.62 (d, J=8.3 Hz, 2H, ArH), 8.21 (s, 1H, H-3). 13C-NMR (126 MHz, CDCl3) δ: 21.6 (CH3), 55.2 (OCH3), 113.2 (CH), 120.7 (C), 128.2 (CH), 128.5 (CH), 129.1 (C), 129.3 (CH), 129.5 (CH), 131.5 (CH), 134.4 (C), 135.4 (C), 137.4 (C), 144.4 (CH), 144.9 (C), 147.2 (C-3), 155.4 (CO), 160.5 (C). MS m/z: 432 (M+, 7.8), 210 (100). Anal. Calcd for C24H20N2O4S·0.25H2O: C, 65.96; H, 4.73; N, 6.41. Found: C, 65.95; H, 4.64; N, 6.46.
7-Methyl-5-[(4-methylphenyl)sulfonyl]-6-phenylimidazo[1,5-a]pyrazin-8(7H)-one (4a)Pale yellow crystals, 16% yield. mp 178–180°C (CHCl3–hexane).17)
5-[(4-Methylphenyl)sulfonyl]-6,7-diphenylimidazo[1,5-a]pyrazin-8(7H)-one (4b)Pale yellow crystals, 10% yield. mp 263–266°C (dec.) (CHCl3–hexane).17)
6-Methyl-5-[(4-methylphenyl)sulfonyl]-7-phenylimidazo[1,5-a]pyrazin-8(7H)-one (4c)A yellow solid, 3% yield. mp 232–235°C (dec.) (CHCl3–hexane).17)
6-(4-Bromophenyl)-7-methyl-5-[(4-methylphenyl)sulfonyl]imidazo[1,5-a]pyrazin-8(7H)-one (4d)White crystals, 20% yield. mp 180–183°C (acetone–hexane).21)
6-(4-Methoxyphenyl)-7-methyl-5-[(4-methylphenyl)sulfonyl]imidazo[1,5-a]pyrazin-8(7H)-one (4e)Ocher crystals, 5% yield. mp 173–176°C (CHCl3–hexane).17)
7-Benzyl-5-[(4-methylphenyl)sulfonyl]-6-phenylimidazo[1,5-a]pyrazin-8(7H)-one (4f)Pale yellow crystals, 36% yield. mp 232–235°C (dec.) (CHCl3–hexane).17)
7-(4-Methoxyphenylmethyl)-5-[(4-methylphenyl)sulfonyl]-6-phenylimidazo[1,5-a]pyrazin-8(7H)-one (4g)Pale yellow crystals, 34% yield. mp 183–185°C (CHCl3–hexane).17)
7-Benzyl-6-(4-methoxyphenyl)-5-[(4-methylphenyl)sulfonyl]imidazo[1,5-a]pyrazin-8(7H)-one (4h)A pale yellow oil, 35% yield.17)
5-[(4-Methylphenyl)sulfonyl]-6-(4-nitrophenyl)-7-phenylimidazo[1,5-a]pyrazin-8(7H)-one (4j)Yellow crystals, 44% yield. mp 244–245°C (CHCl3–hexane).17)
6-(4-Methoxyphenyl)-5-[(4-methylphenyl)sulfonyl]-7-phenylimidazo[1,5-a]pyrazin-8(7H)-one (4k)Pale yellow crystals, 36% yield. mp 253–256°C (dec.) (CHCl3–hexane).17)
General Procedure for Synthesis of Ethyl 5-Oxo-3-phenylpyrazine-2-carboxylates (5)To a stirred solution of ethyl isocyanoacetate (85 mg, 0.75 mmol) in DMF (3 mL) was added DBU (152 mg, 1.00 mmol) at 0°C, and the mixture was stirred for 1 h under atmosphere of oxygen. To the mixture was added 4-trifluoroacetyl-1,3-oxazolium-5-olate 1 (0.50 mmol), and the whole was stirred at 0°C for an additional several hours. After workup with aq. Na2CO3, the mixture was extracted with AcOEt (×3). The combined organic layers were washed with brine, dried over anhyd. Na2SO4, and evaporated. The residue was purified by column chromatography (silica gel, hexane–AcOEt=4 : 1 to 1 : 1) to give the product 5.
Ethyl 4,5-Dihydro-4-methyl-5-oxo-3-phenylpyrazine-2-carboxylate (5a)Yellow crystals, 92% yield. mp 111–113°C (CHCl3–hexane). IR (KBr) cm−1: 2986, 1713, 1671, 1491, 1376, 1317, 1137, 1025, 866, 760, 711. 1H-NMR (500 MHz, CDCl3) δ: 1.05 (t, J=7.0 Hz, 3H, CH2CH3), 3.23 (s, 3H, NCH3), 4.09 (q, J=7.0 Hz, 2H, OCH2), 7.26–7.28 (m, 2H, ArH), 7.52–7.55 (m, 3H, ArH), 8.24 (s, 1H, H-6). 13C-NMR (126 MHz, CDCl3) δ: 13.8 (CH2CH3), 33.5 (NCH3), 61.3 (OCH2), 124.4 (C), 127.9 (CH), 129.0 (CH), 129.9 (CH), 131.9 (C), 146.1 (C-3), 146.2 (C), 156.1 (CO), 164.0 (CO). MS m/z: 258 (M+, 99), 185 (100). HR-MS (EI) for C14H14N2O3: Calcd, 258.1004. Found: 258.1006. Anal. Calcd for C14H14N2O3: C, 65.11; H, 5.46; N, 10.85. Found: C, 65.14; H, 5.43; N, 10.79.
Ethyl 4,5-Dihydro-5-oxo-3,4-diphenylpyrazine-2-carboxylate (5b)Yellow crystals, 88% yield. mp 198–201°C (CHCl3–hexane). IR (KBr) cm−1: 3001, 2983, 1712, 1681, 1569, 1484, 1389, 1378, 1323, 1126, 1004, 928, 754, 710. 1H-NMR (500 MHz, CDCl3) δ: 1.03 (t, J=7.0 Hz, 3H, CH2CH3), 4.11 (q, J=7.0 Hz, 2H, OCH2), 6.96–6.98 (m, 2H, ArH), 7.02–7.07 (m, 2H, ArH), 7.17–7.28 (m, 6H, ArH), 8.36 (s, 1H, H-6). 13C-NMR (126 MHz, CDCl3) δ: 13.8 (CH2CH3), 61.4 (OCH2), 124.5 (C), 127.8 (CH), 128.4 (CH), 129.0 (CH), 129.1 (CH), 129.2 (CH), 129.2 (CH), 131.5 (C), 135.8 (C), 145.7 (C), 147.8 (C-3), 155.7 (CO), 164.3 (CO). MS m/z: 320 (M+, 100). HR-MS (EI) for C19H16N2O3: Calcd, 320.1161. Found: 320.1149. Anal. Calcd for C19H16N2O3·0.33H2O: C, 69.94; H, 5.15; N, 8.59. Found: C, 69.95; H, 4.98; N, 8.41.
Ethyl 4,5-Dihydro-3-methyl-5-oxo-4-phenylpyrazine-2-carboxylate (5c)Yellow crystals, 52% yield. mp 150–153°C (CHCl3–hexane). IR (KBr) cm−1: 3060, 2976, 1705, 1671, 1517, 1385, 1306, 1230, 1095, 776, 709. 1H-NMR (500 MHz, CDCl3) δ: 1.43 (t, J=7.0 Hz, 3H, CH2CH3), 2.40 (s, 3H, 6-CH3), 4.43 (q, J=7.0 Hz, 2H, OCH2), 7.16–7.18 (m, 2H, ArH), 7.52–7.60 (m, 3H, ArH), 8.20 (s, 1H, H-6). 13C-NMR (126 MHz, CDCl3) δ: 14.4 (CH2CH3), 18.2 (CH3), 61.7 (OCH2), 123.7 (C), 127.2 (CH), 129.9 (CH), 130.3 (CH), 136.1 (C), 145.3 (C-3), 146.0 (C), 156.0 (CO), 165.0 (CO). MS m/z: 258 (M+, 100). HR-MS (EI) for C14H14N2O3: Calcd, 258.1004. Found: 258.1006. Anal. Calcd for C14H14N2O3·0.33H2O: C, 63.64; H, 5.59; N, 10.60. Found: C, 63.73; H, 5.42; N, 10.26.
Ethyl 3-(4-Bromophenyl)-4,5-dihydro-4-methyl-5-oxopyrazine-2-carboxylate (5d)Crystals, 85% yield. mp 183–185°C (CHCl3–hexane). IR (KBr) cm−1: 2982, 2901, 1723, 1662, 1485, 1423, 1379, 1313, 1177, 1013, 876, 833, 803, 677. 1H-NMR (500 MHz, CDCl3) δ: 1.12 (t, J=7.0 Hz, 3H, CH2CH3), 3.23 (s, 3H, NCH3), 4.13 (q, J=7.0 Hz, 2H, OCH2), 7.16 (d, J=8.5 Hz, 2H, ArH), 7.69 (d, J=8.5 Hz, 2H, ArH), 8.24 (s, 1H, H-6). 13C-NMR (126 MHz, CDCl3) δ: 13.9 (CH2CH3), 33.5 (NCH3), 61.5 (OCH2), 124.2 (C), 124.5 (C), 129.5 (CH), 130.7 (C), 132.4 (CH), 145.2 (C), 146.5 (C-3), 155.9 (CO), 163.8 (CO). MS m/z: 338 (M+, 55.7), 263 (100). HR-MS (EI) for C14H13BrN2O3: Calcd, 336.0110. Found: 336.0131. Anal. Calcd for C14H13BrN2O3: C, 49.87; H, 3.89; N, 8.31. Found: C, 49.94; H, 3.89; N, 8.55.
Ethyl 4,5-Dihydro-3-(4-methoxyphenyl)-4-methyl-5-oxopyrazine-2-carboxylate (5e)Crystals, 85% yield. mp 148–150°C (CHCl3–hexane). IR (KBr) cm−1: 2983, 2931, 2836, 1714, 1669, 1504, 1315, 1247, 1182, 1029, 871, 816, 694. 1H-NMR (500 MHz, CDCl3) δ: 1.11 (t, J=7.0 Hz, 3H, CH2CH3), 3.25 (s, 3H, NCH3), 3.88 (s, 3H, OCH3), 4.13 (q, J=7.0 Hz, 2H, OCH2), 7.04 (d, J=8.5 Hz, 2H, ArH), 7.19 (d, J=9.0 Hz, 2H, ArH), 8.21 (s, 1H, H-6). 13C-NMR (126 MHz, CDCl3) δ: 13.9 (CH2CH3), 33.4 (NCH3), 55.4 (OCH3), 61.3 (OCH2), 114.5 (CH), 123.8 (C), 124.9 (C), 129.4 (CH), 145.9 (C-3), 146.1 (C), 156.3 (CO), 160.7 (C), 164.2 (CO). MS m/z: 288 (M+, 75.8), 215 (100). HR-MS (EI) for C15H16N2O4: Calcd, 288.1110. Found: 288.1112. Anal. Calcd for C15H16N2O4·0.33H2O: C, 61.23; H, 5.71; N, 9.52. Found: C, 61.24; H, 5.38; N, 9.54.
Ethyl 4-Benzyl-4,5-dihydro-5-oxo-3-phenylpyrazine-2-carboxylate (5f)Yellow crystals, 90% yield. mp 116–119°C (CHCl3–hexane). IR (KBr) cm−1: 3032, 2984, 1715, 1664, 1567, 1519, 1398, 1307, 1197, 1130, 1020, 905, 767, 700. 1H-NMR (500 MHz, CDCl3) δ: 1.01 (t, J=7.0 Hz, 3H, CH2CH3), 4.06 (q, J=7.0 Hz, 2H, OCH2), 5.04 (s, 2H, NCH2Ar), 6.78–6.80 (m, 2H, ArH), 7.03–7.05 (m, 2H, ArH), 7.17–7.19 (m, 3H, ArH), 7.35–7.38 (m, 2H, ArH), 7.46–7.49 (m, 1H, ArH), 8.33 (s, 1H, H-6). 13C-NMR (126 MHz, CDCl3) δ: 13.7 (CH2CH3), 48.7 (NCH2), 61.3 (OCH2), 124.9 (C), 127.1 (CH), 127.8 (CH), 128.4 (CH), 128.5 (CH), 129.9 (CH), 131.0 (C), 135.1 (C), 146.0 (C), 147.0 (C-3), 156.0 (CO), 164.0 (CO). MS m/z: 334 (M+, 82.3), 91 (100). HR-MS (EI) for C20H18N2O3: Calcd, 334.1317. Found: 334.1312. Anal. Calcd for C20H18N2O3: C, 71.84; H, 5.43; N, 8.38. Found: C, 71.54; H, 5.40; N, 8.27.
Ethyl 4,5-Dihydro-4-(4-methoxyphenylmethyl)-5-oxo-3-phenylpyrazine-2-carboxylate (5g)Crystals, 91% yield. mp 75–77°C (CHCl3–hexane). IR (KBr) cm−1: 2966, 2935, 2837, 1723, 1661, 1510, 1297, 1250, 1132, 1028, 805, 764, 712. 1H-NMR (500 MHz, CDCl3) δ: 0.99 (t, J=7.2 Hz, 3H, CH2CH3), 3.74 (s, 3H, OCH3), 4.04 (q, J=7.2 Hz, 2H, OCH2), 4.98 (s, 2H, NCH2Ar), 6.39–6.74 (m, 4H, ArH), 7.07 (d, J=7.2 Hz, 2H, ArH), 7.40 (t, J=7.5 Hz, 2H, ArH), 7.49 (t, J=7.5 Hz, 1H, ArH), 8.29 (s, 1H, H-6). 13C-NMR (126 MHz, CDCl3) δ: 13.7 (CH2CH3), 48.2 (NCH2), 55.2 (OCH3), 61.2 (OCH2), 113.8 (CH), 124.9 (C), 127.2 (C), 128.4 (CH), 128.7 (CH), 128.8 (CH), 129.8 (CH), 131.1 (C), 145.9 (C), 147.0 (C-3), 156.0 (CO), 159.2 (C), 164.0 (CO). MS m/z: 364 (M+, 60.5), 121 (100). HR-MS (EI) for C21H20N2O4: Calcd, 364.1423. Found: 364.1411. Anal. Calcd for C21H20N2O4: C, 69.22; H, 5.53; N, 7.69. Found: C, 68.99; H, 5.42; N, 7.87.
Ethyl 4-Benzyl-4,5-dihydro-3-(4-methoxyphenyl)-5-oxopyrazine-2-carboxylate (5h)Crystals, 91% yield. mp 78–81°C (CHCl3–hexane). IR (KBr) cm−1: 2979, 2843, 1727, 1666, 1609, 1504, 1311, 1250, 1129, 1023, 857, 729, 695. 1H-NMR (500 MHz, CDCl3) δ: 1.07 (t, J=7.1 Hz, 3H, CH2CH3), 3.84 (s, 3H, OCH3), 4.09 (q, J=7.1 Hz, 2H, OCH2), 5.06 (s, 2H, NCH2Ar), 6.83–6.84 (m, 2H, ArH), 6.88 (d, J=8.8 Hz, 2H, ArH), 6.96 (d, J=8.5 Hz, 2H, ArH), 7.20–7.22 (m, 3H, ArH), 8.30 (s, 1H, H-6). 13C-NMR (126 MHz, CDCl3) δ: 13.9 (CH2CH3), 48.6 (NCH2), 55.3 (OCH3), 61.3 (OCH2), 113.8 (CH), 123.1 (C), 125.4 (C), 127.1 (CH), 127.8 (CH), 128.5 (CH), 130.0 (CH), 135.3 (C), 145.9 (C), 146.9 (C-3), 156.2 (CO), 160.7 (C), 164.2 (CO). MS m/z: 364 (M+, 63), 135 (100). HR-MS (EI) for C21H20N2O4: Calcd, 364.1423. Found: 364.1411. Anal. Calcd for C21H20N2O4: C, 69.22; H, 5.53; N, 7.69. Found: C, 69.01; H, 5.60; N, 7.59.
Conversion of 2(1H)-Pyrazinone (5a) to Imidazo[1,5-a]pyrazin-8(7H)-one (6)To a stirred solution of TosMIC (57 mg, 0.290 mmol) in DMF (1 mL) was added DBU (88 mg, 0.581 mmol) at 0°C, and the mixture was stirred for 1 h under atmosphere of argon. To the mixture was added 5a (50 mg, 0.194 mmol), and the whole was stirred at room temperature (r.t.) for an additional 24 h. After workup with aq. Na2CO3, the mixture was extracted with AcOEt (×3). The combined organic layers were washed with brine, dried over anhyd. Na2SO4, and evaporated. The residue was purified by column chromatography (silica gel, hexane–AcOEt=1 : 1) to give ethyl 7,8-dihydro-7-methyl-8-oxo-6-phenylimidazo[1,5-a]pyrazine-5-carboxylate 6 as colorless crystals, 65% yield. mp 163–165°C (CHCl3–hexane). IR (KBr) cm−1: 3146, 2979, 2934, 1709, 1667, 1336, 1321, 1207, 1065, 719. 1H-NMR (500 MHz, CDCl3) δ: 0.78 (t, J=7.2 Hz, 2H, CH2CH3), 3.11 (s, 3H, NCH3), 3.96 (q, J=7.2 Hz, 2H, OCH2), 7.30–7.32 (m, 2H, ArH), 7.49–7.54 (m, 3H, ArH), 8.06 (s, 1H, H-1), 8.83 (s, 1H, H-3). 13C-NMR (126 MHz, CDCl3) δ: 13.2 (CH2CH3), 32.2 (NCH3), 61.5 (OCH2), 109.0 (C), 122.0 (C), 128.8 (CH), 129.6 (CH), 131.4 (CH), 133.2 (CH), 135.0 (CH), 139.2 (C), 155.6 (CO), 161.9 (CO). MS m/z: 297 (M+, 100). HR-MS (EI) for C16H15N3O3: Calcd, 297.1113. Found: 297.1104. Anal. Calcd for C16H15N3O3: C, 64.64; H, 5.09; N, 14.13. Found: C, 64.66; H, 5.07; N, 13.88.
Procedures for Synthesis of 1,3-Thiazolium-5-olates (7a–c)4-Trifluoroacetyl-3-methyl-2-phenyl-1,3-thiazolium-5-olate (7a)To a stirred suspension of N-methyl-N-thiobenzoylglycine (1.00 g, 4.78 mmol) in tert butyl methyl ether (10 mL) was added trifluoroacetic anhydride (TFAA) (2 mL, 14.3 mmol) at 0°C, and the solution was stirred at 0°C for 3 h. After workup with aq. Na2CO3, the mixture was extracted with AcOEt (×3). The combined organic layers were washed with brine, dried over anhyd. Na2SO4, and evaporated. The residue was washed with diethyl ether, and recrystallized from hexane–ethyl acetate to give the product 7a as yellow crystals (1.17 g, 85% yield). mp 131–133°C (AcOEt–hexane). IR (KBr) cm−1: 3062, 3044, 2970, 1673, 1620, 1451, 1396, 1327, 1310, 1261, 1237, 1081, 1049, 1025, 997, 946, 791, 766, 736, 700. 1H-NMR (500 MHz, CDCl3) δ: 4.04 (s, 3H, NCH3), 7.50 (d, J=7.5 Hz, 2H, ArH), 7.59 (t, J=7.5 Hz, 2H, ArH), 7.63–7.66 (m, 1H, ArH). 13C-NMR (126 MHz, CDCl3) δ: 42.5 (NCH3), 115.5 (C), 116.9 (q, 1J=288.5 Hz, CF3), 127.1 (C), 129.3 (CH), 129.7 (CH), 132.6 (CH), 158.5 (C), 168.6 (q, 2J=38.1 Hz, CCF3), 174.2 (C). MS m/z: 287 (M+, 67.5), 121 (100). HR-MS (EI) for C12H8F3NO2S: Calcd, 287.0228. Found: 287.0217.
4-Acetyl-3-methyl-2-phenyl-1,3-thiazolium-5-olate (7b)A mixture of N-methyl-N-thiobenzoylglycine (1.00 g, 4.78 mmol) and acetic anhydride (15 mL) was heated at 120°C for 4 h. After workup with aq. Na2CO3, the mixture was extracted with AcOEt (×3). The combined organic layers were washed with brine, dried over anhyd. Na2SO4, and evaporated. The residue was purified by column chromatography (silica gel, hexane–AcOEt=4 : 1 to 1 : 1) to give the product 7b as yellow crystals (0.903 g, 81% yield). mp 131–132°C (AcOEt–hexane) (mp21) 133–134°C). IR (KBr) cm−1: 3057, 3000, 2920, 1648, 1622, 1375, 1341, 1252, 1173, 1083, 946, 764, 695. 1H-NMR (500 MHz, CDCl3) δ: 2.53 (s, 3H, CCH3), 4.06 (s, 3H, NCH3), 7.46 (dd, J=8.0, 1.5 Hz, 2H, ArH), 7.53–7.60 (m, 3H, ArH). 13C-NMR (126 MHz, CDCl3) δ: 29.7 (CH3), 42.1 (NCH3), 119.0 (C), 127.7 (C), 129.4 (CH), 129.5 (CH), 131.7 (CH), 151.3 (C), 176.5 (C), 187.7 (C). MS m/z: 233 (M+, 36.7), 118 (100). HR-MS (EI) for C12H11NO2S: Calcd, 233.0510. Found: 233.0504.
4-Formyl-3-methyl-2-phenyl-1,3-thiazolium-5-olate (7c)To a mixture of pyridine (0.805 mL, 10 mmol) and DMF (1.925 mL, 25 mmol) in toluene (15 mL) was added phosphoryl chloride (1.2 mL, 12.5 mmol) at 0°C, and the solution was stirred at 0°C for 1 h. To the mixture was added N-methyl-N-thiobenzoylglycine (0.522 g, 2.5 mmol) and the whole was heated at 80°C for an additional 3 h. After workup with aq. Na2CO3, the mixture was extracted with AcOEt (×3). The combined organic layers were washed with brine, dried over anhyd. Na2SO4, and evaporated. The residue was recrystallized from hexane–ethyl acetate to give 7c as amber crystals (0.318 g, 58% yield). mp 160–162°C (AcOEt–hexane). IR (KBr) cm−1: 3056, 3027, 2829, 2782, 2757, 1670, 1604, 1365, 1326, 1307, 1239, 1152, 1051, 859, 774, 759, 697. 1H-NMR (500 MHz, CDCl3) δ: 4.11 (s, 3H, NCH3), 7.47–7.49 (m, 2H, ArH), 7.55–7.63 (m, 3H, ArH), 9.57 (s, 1H, CHO). 13C-NMR (126 MHz, CDCl3) δ: 40.2 (NCH3), 118.1 (C), 127.1 (C), 129.4 (CH), 129.6 (CH), 132.1 (CH), 152.0 (C), 176.2 (C), 178.4 (C). MS m/z: 219 (M+, 30.8), 118 (100). HR-MS (EI) for C11H9NO2S: Calcd, 219.0354. Found: 219.0346.
BiologyAnti-microbial ActivityThe preliminary anti-microbial activies of new compounds were measured in a concentration of 50 mg/L by disc diffusion method.22) Gram-negative bacteria of Acinetobacter baumannii ATC C17978, Escherichia coli TG1, Klebsiella pneumonia IID5209, Pseudomonas aeruginosa 01, Serratia marcescens (clinical isolate), and Vibrio parahaemolyticus RIMD221051 and Gram-positive bacteria of Bacillus subtilis ATC C6633, Enterococcus faecalis IID622, Staphylococcus aureus FDA209P, methicillin-resistant Staphylococcus aureus (MRSA) (clinical isolate), Streptococcus pneumonia IID555, and S. pyogenes 124 were used. Briefly, 100 µL of the middle-logarithmic phase bacteria cells (108 CFU/mL) were spread on surface of the Muller–Hinton agar plate and discs containing 5 µg of new compounds were put on the agar plate, and then incubated at 37°C. The inhibition zone were measured in millimeters at the end of an incubation period of 18–24 h. Dimethyl sulfoxide (DMSO) was used as solvent control and Gentamicin, Kanamycin and Vancomycin were used as standard drugs.
Cytotoxicity DeterminationsA literature procedure was employed when examining the lethal effects of series 3 and 5 compounds on Ca9–22, HSC-2, HSC-3, HSC-4 (purchased from Riken Cell Bank, Tsukuba, Japan), HGF, HPLF and HPC cells (established in Meikai University School of Dentistry according to the guideline of intramural ethic committee) except the time of incubation was extended from 24 to 48 h.30) In brief, different concentrations of test compounds or positive control such as 5-FU (Wako Pure Chemical Industries, Ltd., Osaka, Japan) or melphalan (Sigma-Aldrich Inc., St. Louis, MO, U.S.A.) were incubated at 37°C with the cells in Dulbecco’s modified Eagle’s medium (DMEM) medium supplemented with 10% heat-inactivated fetal bovine serum. The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) method was used in determining cell viability.30)