Regular Articles
Concise and Versatile Synthesis of Sulfoquinovosyl Acyl Glycerol Derivatives for Biological Applications
2017 Volume 65 Issue 6 Pages 566-572
Details
2017 Volume 65 Issue 6 Pages 566-572
Sulfoquinovosyl acylpropanediol (SQAP), a chemically modified analogue of sulfoquinovosyl acylglycerol (SQAG) that occurs in sea algae, has been reported to show a variety of biological activities, including accumulation in tumor cells and the inhibition of tumor cell growth. We report herein on a new concise and versatile synthesis of SQAP itself and derivatives bearing iodoaryl groups and boronclusters. This method should be useful for the design and synthesis of SQAG/SQAP derivatives for diagnosis and the treatment of cancer and related diseases.
The sulfolipid sulfoquinovosyl diacylglycerol (SQDG) produced by higher plants, and other photosynthetic organisms, is characterized by its unique sulfonic acid head group, 6-deoxy-6-sulfo-glucose (sulfoquinovose) and a 1,2-diacyl-sn-glycerol moiety with a long-chain fatty acid.1) Due to its strong amphiphilicity, SQDG would have excellent detergent properties. It has recently been reported that some SQDGs show anti-tumor and anti-viral activity (human immunodeficiency virus (HIV) and herpes simplex virus (HSV)) via the inhibition of DNA polymerase.2–6) Sulfoquinovosyl acylglycerol (SQAG) 1 is one such naturally occurring SQDG derivative that was isolated from sea algae and characterized by Sakaguchi, Sugawara and colleagues7) (Chart 1). It has been reported that SQAG and its analogue sulfoquinovosyl acylpropandiol (SQAP) 2 accumulates in tumor cells and has a low cytotoxicity against normal cells.8) Furthermore, previous structure–activity relationship studies of SQDG/SQAG derivatives indicate that modification of the long alkyl chain moiety on SQDG derivatives with hydrophobic moieties have a negligible effect on their biological activity, particularly their ability to inhibit DNA polymerase and telomerase.9,10) Therefore, SQAP derivatives such as 3 and 4 that contain boron and iodine atoms would be expected to be potent lead compounds for use in cancer therapy and diagnosis for boron-neutron capture therapy11) (BNCT) and for the in vivo imaging of tumor sites,12) respectively (Chart 1).

The total synthesis of SQAP was previously reported by Sugawara and colleagues as shown in Chart 2.8) α-Allyl glycoside α-5 was prepared from D-glucose via α-selective glycosylation and following protection. The introduction of a SO3Na group at the 6-position of α-5 and hydroboration of the allyl group gave the key intermediate α-6, to which a long-chain fatty acid was introduced by a condensation reaction to afford 7, followed by the deprotection of 7 to give the desired compound 2 (SQAP). However, this route is not sufficiently general to permit a wide variety of SQAP derivatives to be prepared, especially when the intermediates or target compounds include functions that are labile under the protection and/or deprotection conditions used in Chart 2 (7 to 2) and other reaction conditions. Specifically, SQAP derivatives bearing NO2, CN, olefin and aryl halide, which would be easily reduced under hydrogenation conditions, would not be synthesized in their synthetic route. As a consequence, the method has only limited use in preparing SQAP analogues.

In this study, we designed a new synthetic route for preparing SQAP and its analogues, 3 and 4. o-Carborane has been frequently used as a hydrophobic pharmacophore for various pharmacologically active compound such as estrogenic agonists13) and vitamin D receptor ligands.14) In addition, compound 3 would be expected to function as a 11B magnetic resonance imaging (MRI) agent, as suggested by Bradshaw et al.15) and us.16,17) SQAP derivatives containing iodine atoms, such as 4, could be utilized as contrast agents for X-ray-computed tomography (CT), which is a well-established tissue imaging technique employed in a variety of research and clinical settings.12,18)
For this purpose, we envisaged a new synthetic route for the preparation of SQAP and its analogues via the thioglucoside 9 and the mesyl derivative 12 (Chart 3). It was assumed that both α- and β-anomers of 6 would be produced by the glycosylation of 9 with mono(p-methoxybenzyl)-protected 1,3-propanediol 10.19) The condensation of 6 with a long chain fatty acid part would give 11, the benzyl groups of which could be removed by hydrogenation to give SQAP derivatives. This synthetic route would be ideal for synthesis of SQAP derivatives bearing base-sensitive functional groups such as o-carborane, whose closo-form (R-C2B10H11) is easily converted to nido-form (R-C2B9H10) by the nucleophilic deboronation reaction.20) Alternatively, it was hypothesized that the conversion of a nucleophile (–OH) of α-6 into a leaving group (–OMs), as shown in 12, would permit acyl moieties to be introduced by SN2 reaction. Namely, we assumed that the mesylation of the primary alcohol of 6 followed by removal of the three benzyl groups would afford 12 and the subsequent SN2 reaction at the mesylate moiety of 12 should afford 13. This new synthetic route would be useful for the synthesis of a wide variety of analogues of SQAP 13, which could not otherwise be obtained using previously reported procedures.

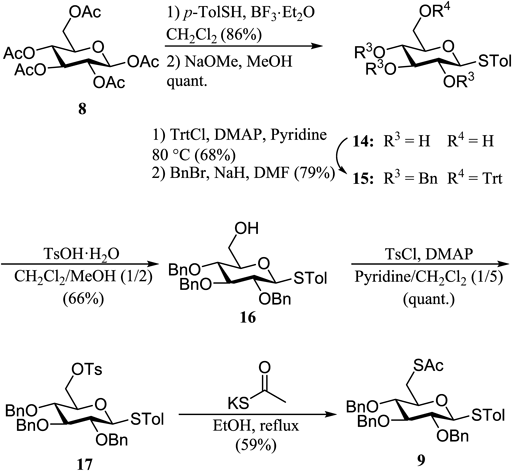
The glycosyl donor 9 was prepared by the glycosylation of commercially available penta-O-acetyl-D-glucopyranose 8, as shown in Chart 4. The β-thioglycoside 14 was prepared by the S-glycosylation of 8 with p-toluenethiol followed by deacetylation using NaOMe in methanol. The OH group at the 6-position of 14 was protected with a trityl group, and the remaining OH groups were protected with benzyl groups to provide 15. The trityl group was then removed by treatment with p-toluenesulfonic acid to give 16,21) which was then converted into 17.22) The 6-OTs group of 17 was substituted with a thioacetyl group to afford 9.
The glycosylation of 9 with 3-(p-methoxybenzyloxy)propanol 10, which was prepared from propanediol,19) afforded 18 as an anomeric mixture (Chart 5). As listed in Table 1, the effect of the solvent used in the reaction on the chemical yield and stereo-selectivity of 18 was investigated. The glycosylation reaction of 9 was initially conducted in CH2Cl2 to afford α-18 in the same yield as β-18 (entry 1). The use of ether-type solvents such as dioxane as a co-solvent improved the α-selectivity, slightly23,24) (entry 2). The addition of tert-butyl methyl ether (TBME) gave better α-selectivity and a combination of NIS and TfOH in a mixture of CH2Cl2 and MeOtBu gave α-18 in reasonable yields and an acceptable α/β ratio (entries 3–5). Although it has reported that α-selectivity is improved by the increasing temperature, chemical yields of this reaction might be decrease probably due to deprotection of PMB group in the presence of TfOH.25)

| Entry | Solvent | Yield (%) | α/β Ratio |
|---|---|---|---|
| 1 | CH2Cl2 | 71 | 50/50 |
| 2 | CH2Cl2/1,4-dioxane (1 : 1) | 65 | 57/43 |
| 3 | CH2Cl2/MeOtBu (1 : 1) | 66 | 67/33 |
| 4 | CH2Cl2/MeOtBu (1 : 2) | 70 | 79/21 |
| 5 | CH2Cl2/MeOtBu (1 : 3) | 62 | 80/20 |
Regarding a o-carborane unit, reaction of decaborane (B10H14) with methyl 10-undecynoate 20,26) which was prepared from 10-undecynoic acid 19, in the presence of 1-butyl-3-methyl imidazolium chloride27) (bmimCl) afforded 21 (Chart 6). Removal of the methyl esters of 21 by hydrolysis with HCl provided the carborane-containing acid 22.28)

The thioacetyl group of α-18 at the 6-position was oxidized by treatment with Oxone® to produce sulfoquinovose derivatives 23 and the p-methoxybenzyl (PMB) group was then removed by DDQ29) to afford the intermediate α-6 (Chart 7). The condensation of the alcohol 6 with stearic acid gave the ester 7, followed by removing the benzyl group by hydrogenation provided 2 (SQAP). In a similar manner, the boron-containing SQAP derivatives 3 was obtained by condensation reaction with 22 followed deprotection of the benzyl groups. However, the use of this route to prepare 4 resulted in the production of deiodinated compounds.

Synthesis of the iodo-containing SQAP 4 is illustrated in Chart 8. The condensation of the 2,3,5-triiodobenzoic acid 25 and tert-butyl 6-aminocaproate 26 gave 27, which after the removal of the tert-butyl ester group, provided 28.30) Mesylation of the OH group of 6 then provided the mesylate derivative 29, followed by removal of the benzyl groups by hydrogenation gave the new intermediate 12, to which 28 was introduced by substitution reaction31) to give the iodo-containing derivative 4. Using this synthetic route and reaction conditions, it is possible to access various SQAP derivatives that contain labile functional groups.

In this study, we report on new synthetic routes for preparing SQAP itself and its derivatives 3 and 4. The intermediate α-6 was prepared from 8 via the thioglycoside donor 9, whose glycosylation with mono(PMB)-protected propandiol in a mixture of CH2Cl2 and MTBE (1 : 3) afforded the O-glycoside 18 in good chemical yield with a high degree of α selectivity. Subsequent condensation reaction and debenzylation provided SQAP and its derivative 3. This synthetic route would be ideal for synthesis of 3, whose o-carborane moieties are decomposed by strong base. Furthermore, the conversion of the nucleophile (–OH) of α-6 into a leaving group (–OMs) enabled various SQAP derivatives that contain labile functional groups, such as 4, to be prepared. These new synthetic routes for preparing SQAP derivatives might be useful for the design and synthesis of chemically modified SQAG/SQAP derivatives, which have the potential for use in the therapy and diagnosis of cancer and related diseases.
IR spectra were recorded on Perkin-Elmer FT-IR spectrophotometer at room temperature. 1H- (300 MHz) and 13C- (75 MHz) NMR spectra were recorded on a JEOL Always 300 spectrometer. 1H- (400 MHz) and 11B- (128 MHz) NMR spectra were recorded on a JEOL Lambda 400 spectrometer. 13C- (150 MHz) NMR spectra were recorded on a JEOL ECA-600 spectrometer. Tetramethylsilane was used as an internal reference for 1H- and 13C-NMR measurements in CDCl3 and CD3OD. The sodium salt of 3-(trimethylsilyl)propionic-2,2,3,3-d4 acid (TSP) was used as an external reference for the 1H-NMR and 1,4-dioxane for 13C-NMR measurements, which were conducted in D2O. 11B-NMR spectra were measured in a quartz NMR tube using boron trifluoride diethyl ether complex in CDCl3 as an external reference (0 ppm). Elemental analyses were performed on a Perkin-Elmer CHN 2400 analyzer. MS spectra were recorded on a JEOL JMS-SX102A and Agilent (Varian) 910-MS. Thin-layer (TLC) and silica gel column chromatographies were performed using a Merck 5554 (silica gel) TLC plates and Fuji Silysia Chemical FL-100D, respectively. Oxone® (potassium peroxymonosulfate, KHSO5·0.5KHSO4·0.5K2SO4) was purchased from Sigma-Aldrich.
β-1-p-Tolylthio-2,3,4-tri-O-benzyl-6-thioacetyl-D-glucopyranose (9)Potassium thioacetate (1.51 g, 13 mmol) was added to a suspension of 1722) (4.70 g, 6.54 mmol) in ethanol (75 mL) at room temperature, and the resulting mixture was stirred at reflux temperature for 5 h. After cooling the reaction solution to room temperature, the mixture was evaporated under reduced pressure, and the resulting residue was dissolved in AcOEt, washed with H2O and brine, dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (hexanes–AcOEt=4 : 1) and recrystallized from hexanes to give 9 (2.40 g, 3.91 mmol, 60%) as colorless needles; mp 79.5–79.9°C. 1H-NMR (300 MHz, CDCl3, TMS): δ=2.36 (s, 6H), 3.01–3.04 (dd, 1H, J=6.9, 13.5 Hz), 3.35–3.48 (m, 3H), 3.53 (dd, 1H, J=2.4, 13.8 Hz), 3.68 (t, 1H, J=8.7 Hz), 4.55 (d, 1H, J=9.9 Hz), 4.64 (d, 1H, J=10.8 Hz), 4.73 (d, 1H, J=10.2 Hz), 4.82–4.93 (m, 4H), 7.132 (d, 2H, J=7.8 Hz), 7.28–7.42 (m, 15H), 7.47 (d, 2H, J=8.1 Hz) ppm. 13C-NMR (75 MHz, CDCl3): δ=21.0, 30.4, 30.9, 75.1, 75.3, 75.7, 77.7, 80.1, 80.8, 86.4, 87.6, 127.7, 127.7, 127.9, 128.0, 128.1, 128.3, 128.3, 128.4, 129.4, 129.5, 132.8, 137.6, 137.7, 137.9, 138.1, 194.6 ppm. IR (ATR): 3091, 3065, 3032, 2907, 2869, 1946, 1872, 1811, 1696, 1607, 15589, 1495, 1455, 1398, 1354, 1273, 1252, 1213, 1201, 1132, 1104, 10069, 1029, 992, 968, 909, 847, 805, 797, 738, 695, 657, 626, 578, 559, 496, 487 cm−1. High resolution (HR)-MS (FAB+): Calcd for [M+Na]+, C36H38O5S2, 637.2051. Found, 637.2051. Elemental analysis Calcd (%) for C36H38O5S2: C, 70.33; H, 6.23. Found: C, 70.55; H, 6.13.
1-O-(2,3,4-Tri-O-benzyl-6-thioacetyl-α-D-glucopyranosyl)-3-O-p-methoxybenzylpropane-1,3-diol (18)NIS (562 mg) was added to a mixture of 9 (307 mg, 0.50 mmol), 3-(4-methoxybenyloxy)-1-propanol 1021) (182 mg, 0.93 mmol), molecular sieves (4 Å, 1.0 g) in freshly distilled CH2Cl2 (3.3 mL) and tert-butyl methyl ether (6.7 mL) at −40°C. After 15 min, TfOH (cat.) was added, and the reaction mixture was gradually warmed to −30°C. After 2 h, the reaction was quenched by adding Et3N and the insoluble material was removed by filtration. The filtrate was diluted with CHCl3 and washed with 10% Na2S2O3 aq., 1 N HCl aq. and brine, dried over Na2SO4 and filtered. The filtrate was concentrated under reduced pressure and the remaining residue was purified by silica gel column chromatography (hexanes–AcOEt=10 : 1) and HPLC (Senshu Pak PEGASIL-B 20ϕ×250 nm, MeOH/H2O (0.1% TFA)=90/10, flaw rate 10 mL/min) to give α-18 (215 mg, 63%) and β-18 (47.8 mg, 14%) as colorless syrups.
α-18: 1H-NMR (300 MHz, CDCl3, TMS): δ=1.92 (m, 2H), 2.29 (s, 3H), 3.05 (dd, 1H, J=7.7 Hz), 3.28–3.61 (m, 6H), 3.72–3.82 (m, 5H), 3.96 (t, 1H, J=9.2 Hz), 4.42 (s, 2H), 4.60–4.64 (m, 2H), 4.68 (d, 1H, J=3.6 Hz), 4.74 (d, 1H, J=12.0 Hz), 4.80 (d, 1H, J=10.5 Hz), 4.89 (d, 1H, J=10.5 Hz), 4.98 (d, 1H, J=10.5 Hz), 6.84–6.88 (m, 2H), 7.23–7.36 (m, 15H) ppm. 13C-NMR (75 MHz, CDCl3): δ=29.5, 30.2, 30.7, 54.9, 64.8, 66.6, 69.1, 72.4, 72.8, 75.0, 75.4, 76.6, 77.0, 77.2, 77.4, 79.9, 80.3, 81.5, 96.4, 113.5, 127.4, 127.7, 127.7, 128.0, 129.0, 130.0, 137.7, 137.9, 138.5, 158.9, 194.6 ppm. IR (ATR): 3089, 3063, 3031, 3005, 2911, 2864, 1692, 1613, 1586, 1513, 1498, 1455, 1400, 1357, 1303, 0247, 1209, 1067, 1028, 953, 911, 820, 734, 696, 628 cm−1. HR-MS (FAB+): Calcd for [M+Na]+, C40H46O8S, 709.2811. Found, 709.2814.
β-18: 1H-NMR (300 MHz, CDCl3, TMS): δ=1.92 (m, 2H), 2.29 (s, 3H), 2.94 (dd, 1H, J=13.6 Hz), 3.32–3.42 (m, 3H), 3.51–3.69 (m, 5H), 3.77 (s, 3H), 3.95–4.00 (m, 1H), 4.33 (d, 1H, J=8.0 Hz), 4.40 (s, 2H), 4.64 (d, 1H, J=12.4 Hz), 4.70 (d, 1H, J=10.0 Hz), 4.76 (d, 1H, J=11.2 Hz), 4.85–4.93 (m, 3H) ppm. 13C-NMR (75 MHz, CDCl3): δ=30.2, 30.5, 31.0, 55.2, 66.8, 67.0, 72.6, 73.8, 74.7, 75.1, 75.7, 80.3, 82.2, 84.4, 103.4, 113.7, 127.6, 127.9, 127.9, 128.0, 128.2, 128.3, 128.3, 128.4, 129.1, 130.5, 137.7, 138.3, 138.4, 159.1, 194.9 ppm. IR (ATR): 3089, 3063, 3031, 3005, 2911, 2864, 1692, 1613, 1586, 1513, 1498, 1455, 1400, 1357, 1303, 0247, 1209, 1067, 1028, 953, 911, 820, 734, 696, 628 cm−1. HR-MS (FAB+): Calcd for [M+Na]+, C40H46O8S, 709.2811. Found, 709.2813.
Methyl 1-O-1,2-Dicarbadodecaborane-dodecanoate (21)To a solution of B10H14 (122 mg, 1.00 mmol) in toluene (5.4 mL), 2026) (245 mg, 1.25 mmol, prepared from 19 as indicated in Chart 6 in this work) and 1-butyl-3-methylimidazolium chloride (bmimCl) (59.4 mg, 0.34 mmol) were added. The resulting solution was stirred at reflux condition for 3 h. After evaporation, the resulting residue was purified by silica gel column chromatography (hexanes–AcOEt=10 : 1) to afford 21 as a colorless amorphous (65.8 mg, 21% yield).: 1H-NMR (300 MHz, CDCl3, TMS): δ=1.25–1.63 (m, 19H), 2.15–2.21 (m, 2H), 2.30 (t, 2H, J=7.5 Hz), 3.49 (s, 3H), 3.56 (br, 1H) ppm. 13C-NMR (75 MHz, CDCl3): δ=24.7, 28.7, 28.8, 28.9, 28.9, 29.1, 33.9, 38.0, 51.4, 60.9, 75.4 174.1 ppm. 11B{1H}-NMR (128 MHz, CDCl3, BF3·OEt2): δ=−13.2 (s, 2B), −12.3 (s, 2B), −11.4 (s, 2B), −9.4 (s, 2B), −6.0 (s, 1B), −2.5 (s, 1B) ppm. IR (ATR): 3059, 2956, 2924, 2855, 2605, 2568, 1732, 1467, 1435, 1416, 1383, 1362, 1333, 1297, 1258, 1217, 1189, 1173, 1136, 1106, 1075, 1044, 1017, 998, 975, 930, 888, 855, 799, 721, 660 cm−1. HR-MS (FAB-): Calcd for [M−H], C12H30B10O2, 316.3176. Found, 316.3188.
1-O-1,2-Dicarbadodecaborane-dodecanoic Acid (22)A suspension of 21 (65.8 mg, 0.209 mmol) in aqueous solution of 5 N HCl (3.5 mL) was stirred at reflux temperature for 24 h. After cooling the reaction solution to room temperature, the mixture was dissolved in AcOEt, washed with 2 N HCl solution and brine, dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (hexanes–AcOEt=4 : 1) to give 22 (36.9 mg, 0.123 mmol, 59%) as colorless amorphous. 1H-NMR (300 MHz, CDCl3, TMS): δ=1.26–1.65 (m, 19H), 2.16–2.21 (m, 2H), 2.35 (t, 2H, J=7.5 Hz), 3.56 (br, 1H) ppm. 13C-NMR (75 MHz, CDCl3): δ=24.5, 28.8, 28.8, 29.0, 29.1, 33.9, 38.1, 60.9, 75.4, 179.5 ppm. 11B{1H}-NMR (128 MHz, CDCl3, BF3·OEt2): δ=−13.1 (s, 2B), −12.3 (s, 2B), −11.4 (s, 2B), −9.4 (s, 2B), −5.9 (s, 1B), −2.5 (s, 1B) ppm. IR (ATR): 3063, 2934, 2857, 2570, 1697, 1469, 1432, 1411, 1330, 1286, 1248, 1228, 1209, 1124, 1069, 1020 cm−1. HR-MS (FAB-): Calcd for [M−H]−, C11H28B10O2, 302.3020. Found, 302.2978.
Sodium 1-O-(2,3,4-Tri-O-benzyl-6-sulfo-α-D-quinovopyranosyl)-3-O-p-methoxybenzyl-propane-1,3-diol (23)Oxone® was added to a suspension of α-18 (161 mg, 0.23 mmol) and sodium acetate (390 mg, 0.48 mmol) in acetic acid (3.4 mL) and the resulting mixture was stirred at room temperature for 24 h. The mixture was then diluted with AcOEt and washed with saturated NaHCO3 aq., and brine, dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (CHCl3–MeOH=4 : 1) to give 23 (146 mg, 87%) as a colorless amorphous. 1H-NMR (300 MHz, CDCl3, TMS): δ=1.837 (m, 2H), 3.17–3.31 (m, 2H), 3.37–3.51 (m, 5H), 3.61 (s, 3H), 3.87–3.98 (m, 2H), 4.26 (t, 1H, J=9.0 Hz), 4.32 (s, 2H), 4.52–4.56 (m, 3H), 4.64 (d, 1H, J=11.1 Hz), 4.78 (d, 1H, J=11.1 Hz), 4.87 (d, 1H, J=11.1 Hz), 4.94 (d, 1H, J=3.0 Hz), 5.30 (s, 2H), 6.74 (d, 2H, J=8.7 Hz), 7.13–7.25 (m, 15H) ppm. 13C-NMR (75 MHz, CDCl3): δ=20.8, 29.3, 52.0, 55.0, 55.1, 65.1, 66.3, 67.4, 72.3, 72.5, 75.0, 75.4, 77.3, 80.2, 80.5, 81.7, 96.4, 113.7, 127.4, 127.7, 127.7, 127.7, 128.2, 128.3, 128.3, 129.5, 129.9, 130.1, 138.1, 138.3, 138.8, 159.1, 175.3 ppm. IR (ATR): 3461, 3064, 3032, 3004, 2933, 2866, 1728, 1612, 1587, 1513, 1498, 1454, 1401, 1363, 1330, 1302, 1246, 1211, 1175, 1086, 1047, 1027, 996, 911, 886, 821, 796, 734, 696, 675, 665, 636, 607, 573, 550, 532, 463 cm−1. HR-MS (FAB-): Calcd for [M−Na]−, C38H43O10S, 691.2582. Found, 691.2574.
Sodium 1-O-(2,3,4-Tri-O-benzyl-6-sulfo-α-D-quinovopyranosyl)propane-1,3-diol (6)DDQ (164 mg, 0.72 mmol) was added to a suspension of 24 (413 mg, 0.60 mmol) in CH2Cl2 (30 mL) and H2O (1.6 mL) and the resulting mixture was stirred at room temperature for 90 min. It was then extracted with CHCl3 and washed with 2 N HCl aq. and brine, dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (CHCl3–MeOH=8 : 1 to 6 : 1) to give 6 (314 mg, 0.529 mmol, 88%) as a pale brown solid. Spectroscopic data (1H- and 13C-NMR spectra) for 6 were consistent with the previously reported data.32)
Sodium 3-O-(2,3,4-Benzyl-6-sulfo-α-D-quinovopyranosyl)-1-O-1,2-dicarbadodecaborane-1-dodecanoyl-propane-1,3-diol (24)6 was added to a solution of (EDC·HCl) (49.2 mg, 0.26 mmol), DMAP (2.1 mg, 17 µmol) and 22 (41.7 mg, 0.17 mmol) in CH2Cl2 (6.0 mL) and the resulting mixture was stirred at room temperature for 24 h. The mixture was then diluted with CHCl3, washed with 2 N HCl aq. and brine, dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (CHCl3–MeOH=4 : 1, containing 1% (v/v) AcOH) to give 24 (125.2 mg, 85%) as a colorless amorphous. 1H-NMR (300 MHz, CDCl3, TMS): δ=1.15 (m, 10H), 2.00–2.23 (m, 19H), 2.55 (t, J=6.6 Hz, 2H), 2.76 (s, 2H), 3.22–3.53 (m, 2H), 3.51 (br s, 2H), 3.95 (t, 2H, J=8.7 Hz), 4.16–4.22 (m, 3H), 4.54–4.74 (m, 4H), 4.83–4.96 (m, 3H) ppm. 13C-NMR (150 MHz, CDCl3, TMS): δ=24.7, 28.4, 28.8, 29.0, 29.0, 29.2, 29.7, 31.9, 34.4, 37.9, 52.2, 52.3, 60.4, 61.0, 63.6, 65.0, 67.0, 73.0, 74.9, 75.3, 75.5, 76.8, 77.0, 77.2, 80.0, 80.2, 81.8, 96.2, 127.6, 127.7, 127.8, 127.9, 128.4, 137.7, 138.0, 138.5, 175.9 ppm. 11B{1H}-NMR (128 MHz, CDCl3, BF3·OEt2): δ=−11.6 (s, 6B), −9.5 (s, 2B), −5.9 (s, 1B), −2.5 (s, 1B) ppm. IR (ATR): 3434, 3063, 2929, 2858, 2858, 2583, 1728, 1498, 1455, 1360, 1160, 1055, 1029, 734, 697, 633, 543 cm−1. HR-MS (ESI-): Calcd for [M−Na]−, C41H61B10O10S−, 855.4921. Found, 855.4969.
Sodium 3-O-(6-Sulfo-α-D-quinovopyranosyl)-1-O-1,2-dicarbadodecaborane-1-pentanoyl-propane-1,3-diol (3)5% Pd/C (820 mg) was added to a solution of 24 (75.3 mg, 0.12 mmol) in MeOH (6.0 mL) under an atmosphere of argon and the mixture was stirred at room temperature for 24 h under an atmosphere of H2 gas. The resulting solution was then filtered through a pad of celite and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (CHCl3–MeOH=4 : 1 to 3 : 1) to give 3 (24.9 mg, 48%) as a colorless amorphous. 1H-NMR (300 MHz, CD3OD, TMS): δ=1.15 (m, 10H), 2.00–2.23 (m, 19H), 2.55 (t, J=6.6 Hz, 2H), 2.76 (s, 2H), 3.22–3.53 (m, 2H), 3.51 (br s, 2H), 3.95 (t, 2H, J=8.7 Hz), 4.16–4.22 (m, 3H), 4.54–4.74 (m, 4H), 4.83–4.96 (m, 3H) ppm. 13C-NMR (150 MHz, CD3OD, TMS): δ=26.1, 29.8, 30.2, 30.3, 30.4, 30.6, 30.6, 30.7, 31.0, 33.0, 35.1, 54.3, 63.1, 65.4, 69.6, 73.5, 75.0, 75.0, 99.6, 176.0 ppm. 11B{1H}-NMR (128 MHz, CD3OD, BF3·OEt2): δ=−12.4–10.9 (m, 3B), −9.0 (s, 2B), −5.6 (s, 1B), −2.3 (s, 1B), 32.4 (s, 3B) ppm. IR (ATR): 3351, 2925, 2855, 1715, 1353, 1164, 1030 cm−1. HR-MS (ESI-): Calcd for [M−Na]−, C20H43B10O10S, 585.3513. Found, 585.3520.
Sodium 3-O-(2,3,4-Tri-O-benzyl-6-sulfo-α-D-quinovopyranosyl)-1-O-mesylpropane-1,3-diol (29)To a solution of 6 (100 mg, 0.17 mmol) and DMAP (10.3 mg, 84 µmol) in anhydrous pyridine (2.5 mL), methanesulfonyl chloride (MsCl) (77.0 mg, 0.67 mmol) was added at 0°C, and the mixture was stirred at room temperature. After 5 h, methanol (4.0 mL) was added, and the reaction mixture was extracted with chloroform, washed with 2 N HCl aq. and brine, dried over Na2SO4, filtered and evaporated under reduced pressure. The residue was purified by silica gel column chromatography (CHCl3–MeOH=25 : 1 containing 1% (v/v) AcOH) to give 29 (71.2 mg, 63%) as a colorless amorphous. mp 164–165. 1H-NMR (300 MHz, CDCl3, TMS): δ=1.99 (br s, 2H), 2.85 (s, 3H), 3.16–3.56 (m, 5H), 3.88–3.99 (m, 2H), 4.14 (t, 1H, J=9.6 Hz), 4.29 (br s, 2H), 4.52–4.69 (m, 4H), 4.78 (d, 1H, J=10.2 Hz), 4.90 (d, 1H, J=10.8 Hz), 4.97 (br s, 1H) ppm. 13C-NMR (75 MHz, CDCl3): δ=14.1, 28.9, 29.7, 36.7, 52.1, 63.5, 67.4, 68.1, 72.8, 75.0, 75.4, 77.2, 80.0, 80.7, 81.5, 96.2, 127.5, 127.7, 127.8, 128.3, 128.4, 137.9, 138.1, 138.6 ppm. IR (ATR): 3441, 3032, 2931, 1728, 1605, 1562, 1498, 1455, 1406, 1351, 1290, 1214, 1170, 1116, 1087, 1046, 1027, 975, 941, 837, 790, 735, 696, 674, 635, 579, 527, 461 cm−1. HR-MS (FAB-): Calcd for [M]−, C31H37O11S2, 649.1777. Found, 649.1775.
Sodium 3-O-(6-Sulfo-α-D-quinovopyranosyl)-1-O-mesylpropane-1,3-diol (12)5% Pd/C (575 mg) was added to a solution of 29 (59.3 mg, 88 µmol) in MeOH (6.0 mL) under an atmosphere of argon and the mixture was stirred at room temperature for 16 h under an atmosphere of H2 gas. It was then filtered through a pad of celite and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (CHCl3–MeOH=5 : 1 to 4 : 1) to give 12 (24.8 mg, 70%) as a colorless amorphous. 1H-NMR (300 MHz, CD3OD, TMS): δ=1.99–2.16 (m, 2H), .2.91 (dd, 1H, J=9.2, 14.3 Hz), 3.04–3.10 (m, 4H), 3.32–3.43 (m, 2H), 3.48–3.67 (m, 2H), 4.03–4.12 (m, 2H), 4.35–4.48 (m, 2H), 4.76 (d, 1H, J=3.6 Hz) ppm. 13C-NMR (75 MHz, CD3OD): δ=30.3, 37.1, 54.3, 64.8, 69.5, 69.7, 73.5, 75.0, 75.1, 99.7 ppm. IR (ATR): 3383, 3021, 2933, 1722, 1639, 1413, 1338, 1165, 1031, 979, 938, 846, 829, 790, 755, 724, 699, 664, 628, 590, 526 cm−1. HR-MS (FAB-): Calcd for [M]−, C10H19O11S2, 379.0369. Found, 379.0368.
Sodium 3-O-(6-Sulfo-α-D-quinovopyranosyl)-1-O-(6-(2,3,5-triiodobenzoyl)-aminohexanoyl)-propane-1,3-diol (4)To a suspension of 12 (24.8 mg, 62 µmol) and 6-(2,3,5-triiodobenzoyl)aminohexanoic acid 28 (170 mg, 0.28 mmol) in anhydrous DMF (1.5 mL), CsF (93.6 mg, 0.62 mmol) and NaI (36.9 mg, 0.25 mmol) was added at room temperature, and the resulting mixture was stirred at 70°C. After 48 h, methanol (1.0 mL) was added, and the reaction mixture was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (CHCl3–MeOH=6 : 1 to 4 : 1 to 3 : 1) and HPLC (Senshu Pak PEGASIL-B 20ϕ×250 nm, CH3CN–H2O (0.1% TFA)=78 : 22, flaw rate 10 mL/min) to give 4 (24.1 mg, 44%) as a colorless amorphous. mp 169–170°C. 1H-NMR (300 MHz, CD3OD, TMS): δ=1.40–1.50 (m, 2H), 1.59–1.72 (m, 4H), 1.93–2.01 (m, 2H), 2.37 (t, 2H, J=7.4 Hz), 2.91 (dd, J=9.2, 14.3 Hz), 3.07 (t, 1H, J=9.3 Hz), 3.35–3.41 (m, 4H), 3.45–3.52 (m, 1H), 3.64 (t, 1H, J=9.3 Hz), 4.02–4.10 (m, 2H), 4.17–4.31 (m, 2H), 4.74 (d, 1H, J=4.6 Hz), 7.54 (d, 1H, J=2.4 Hz), 8.29 (d, 1H, J=1.8 Hz) ppm. 13C-NMR (75 MHz, CDCl3): δ=25.6, 27.6, 29.6, 34.7, 40.7, 66.0, 73.6, 75.1, 94.7, 99.6, 107.2, 112.8, 136.1, 148.5 ppm. IR (ATR): 3260, 2928, 2860, 1725, 1631, 4556, 1520, 1436, 1416, 1392, 1359, 1300, 1152, 1110, 1089, 1050, 1028, 1012, 910, 890, 864, 828, 788, 738, 699, 662, 626, 585, 520, 504 cm−1. HR-MS (FAB-): Calcd for [M−Na]−, C22H29I3NO11S, 895.8595. Found, 895.8597.
This work was supported by Grants-in-Aid from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan (No. 19659026, 22390005, and 24659085 for S.A. and No. 25893259 for T. T.) and TUS (Tokyo University of Science) fund for strategic research areas. We wish to express our appreciation to Prof. Kengo Sakaguchi (Research Institute of Science and Technology, Tokyo University of Science) and Prof. Fumio Sugawara (Department of Applied Biological Science, Faculty of Science and Technology, Tokyo University of Science) for providing SQAP as a reference. We also wish to express our appreciation for the aid provided by Mrs. Fukiko Hasegawa (Faculty of Pharmaceutical Sciences, Tokyo University of Science) for mass spectral measurements.
The authors declare no conflict of interest.