Current Topics : Reviews
Peptide-Based Cancer-Targeted DDS and Molecular Imaging
2017 Volume 65 Issue 7 Pages 618-624
Details
2017 Volume 65 Issue 7 Pages 618-624
Targeting cancer cell-surface receptors is an attractive approach for cancer treatment and diagnosis. Peptides having high binding affinities to receptors overexpressed in cancer cells are useful because of their simple structure, low immunogenicity, and easy, cost-effective chemical synthesis. A number of peptide ligands have been developed for cancer cell-surface receptors and applied to nanoparticles with anticancer drugs, genes, small interfering RNAs (siRNAs), and molecular imaging agents. In particular, recent findings have revealed that peptide-modified PEGylated liposome-encapsulated drugs are effective in cancer-targeted therapy and cancer cell-specific imaging. This review discusses peptide-modified nanoparticles for drug delivery systems (DDS) and molecular imaging, focusing on peptide ligands for somatostatin receptors, integrin, transferrin receptor, human epidermal growth factor 2 (HER2), etc. In addition, methods to improve binding affinity or endosomal escape with spacer peptides and stimuli (internal and external) are discussed.
The number of cancer patients has been increasing, owing to the aging of populations as a result of advances in medical technology. As treatment, surgical excision, chemotherapy, radiation therapy, and hormone therapy are typically used alone or in combination depending on the type and stage of cancer. However, tumor cells have heterogenetic properties in terms of gene expression, morphology, proliferation, metabolic activity, metabolism, motility, and metastatic potential.1) Therefore, it is difficult to achieve sufficient therapeutic effects using the above treatments. Recently, molecularly targeted drugs inhibiting signaling pathways related to tumor cell growth and survival have been developed for clinical use.2) Targeted therapy is a new approach that enables selective cancer treatment.3,4)
For cancer targeting, a desired receptor is overexpressed in tumor cells or tumor-related blood vessels but not in normal cells.5) Many cell-surface receptors have been found in cancer cells, for example, integrin receptors as extracellular matrixadhesion molecules,6) epidermal growth factor receptors (EGFRs) and human epidermal growth factor 2 (HER2) as tyrosine kinase receptors,7,8) somatostatin receptors as G-protein-coupled receptors (GPCRs), and luteinizing hormone-releasing hormone (LHRH) receptor as a hormone receptor.9) Selective ligands binding to these receptors have been developed based on the structure of chemotherapeutic molecules, carbohydrates, peptides, and antibodies.10–12) The application of the ligands to anticancer drugs is a rational approach for drug delivery systems (DDS) by directly conjugating to small molecular drugs or formulating nanoparticle-encapsulated small molecular drugs, nucleic acids, peptides, and proteins.13,14) In addition, this approach is effective in molecular imaging that enables us to visualize tumor cells or their microenvironments in positron-emission tomography (PET), single photon-emission computed tomography (SPECT), magnetic resonance imaging (MRI), and optical imaging.14)
Among ligands, peptides obtained from endogenous protein sequences are relatively safe and show low immunogenicity, in addition to structural simplicity. Peptides are also much less costly compared with monoclonal antibodies that are large protein ligands,15) because peptides can be readily prepared on a large scale with progress in chemical synthesis methods.16) Therefore, peptides are expected to be effective, selective ligands for disease treatment.
Peptide-modified nanoparticles such as liposomes, polymeric micelles, and nanogels have attracted attention for cancer treatment and diagnosis.13,17) Nanoparticles (<200 nm) are nonspecifically leaked to tumor cells due to the enhanced permeability and retention (EPR) effect occurring with increasing tumor vascular permeability.18–20) The modification of nanoparticles with peptide ligands enables their specific accumulation in target organs and tumor cells by receptor-mediated uptake such as endocytosis.21,22) In addition, the physicochemical properties of their surface such as hydrophobicity and hydrophilicity also affect the biodistribution of nanoparticles. Nanoparticles with hydrophobic surfaces interact with blood components and are subsequently cleared by the reticuloendothelial system (RES).23) To avoid uptake by the RES and remain in the blood circulation, a hydrophilic poly(ethylene glycol) (PEG) is generally modified for nanoparticles to provide steric stabilization effects, and several types of PEGylated nanoparticles are used as drug carriers and imaging probes.24,25) In addition, some peptides have antimicrobial or cell-penetrating activity. For intracellular drug delivery, cell-penetrating peptides (CPPs) are useful for translocating small and macromolecules inside cells.26) CPPs are short peptides obtained from synthetic peptide and membrane transduction domains, etc. The transactivator of transcription (TAT) protein of the human immunodeficiency virus (HIV) is known to bring various molecules into the cell cytoplasm nonspecifically.27,28) TAT-modified liposomes enable gene transfection more easily compared with the nonmodified lyposomes.29) Polyarginine 8 (R8) also shows cell-penetrating activity, and R8-modified liposomes can deliver small interfering RNA (siRNA) into lung cancer cell lines.30) In the absence of specificity for tumor cells, the combination of CPPs and tumor-specific ligands including peptides is effective in targeted delivery.
Thus, there are many types of peptide ligand expected to be applied to cancer therapy. Here, the focus is on nanoparticles modified with peptide ligands for targeted delivery to cancer cells and their applications in DDS and imaging.
Peptides are fragments of proteins but they can function as well as parent full-length proteins with the application of molecular design technologies. After the discovery of hormones, it was found that relatively short peptides had biological activities similar to those of native hormones. Therefore, several drugs based on peptides were developed as lead compounds.31) In the discovery of tumor-related peptides, phage-display peptide libraries greatly contributed to identifying effective sequences as ligands.32) In addition, the synthesis and modification of peptides were made easier and simpler with advances in chemical synthesis, solid-phase synthesis, and chemical ligation technologies.33,34) Although the stability of peptides in vivo is a problem, incorporating unnatural amino acids such as D-amino acids or cyclic structures can prevent peptide degradation.35,36) Table 1 summarizes peptide ligands for drug delivery and molecular imaging.
| Peptide ligand | Target receptor | Target cancer cells | Carrier | Drug/imaging modality | Refs. |
|---|---|---|---|---|---|
| Octreotide | SSTR2 | Breast | Liposome | Cantharidin/— | 47) |
| Octreotide | SSTR2 | Lung | Liposome | DOX/fluorescence | 48) |
| Octreotide | SSTR2 | Glioblastoma | Liposome | —/dual imaging (PET/MRI) | 49) |
| RGD | αvβ3 integrin | Melanoma | Liposome | DOX/fluorescence | 56) |
| cyclic RGD | αvβ3 integrin | Melanoma, breast, colon | Liposome | Matrine/— | 57) |
| cyclic RGD | αvβ3 integrin | Colon | Liposome | DOX/fluorescence | 58) |
| cyclic RGD | αvβ3 integrin | Glioblastoma | QD | —/dual imaging (PET/NIR fluorescenxce) | 65) |
| ATN-161 | α1β5 integrin | Breast | Liposome | DOX/fluorescence | 67) |
| T7 | TFR | Ovarian | Liposome | PTX/fluorescence | 80) |
| T7/TAT | TFR | Glioma | Liposome | DOX/NIR fluorescenxce | 81) |
| T7/TAT | TFR | Lung | Liposome | PTX/NIR fluorescenxce | 82) |
| KCCYSL | HER2 | Breast | Liposome | DOX/— | 91) |
| LTVSPWY | HER2 | Ovarian | Magnetic nanoparticle | —/MRI | 93) |
| AHNP | HER2 | Breast | Liposome | DOX/— | 95) |
| LHRL | LHRH receptor | Lung | Polymer, dendrimer, liposome | PTX/NIR fluorescenxce | 101) |
| NGR | Aminopeptidase N | Fibrosarcoma | Liposome | siRNA and DOX/— | 103) |
| NGR | Aminopeptidase N | Fibrosarcoma | Micelle | DTX/fluorescence | 104) |
| NGR/heat activatable CPP | Aminopeptidase N | Fibrosarcoma | Liposome | DOX/fluorescence | 105) |
| NGR | Aminopeptidase N | Glioma | QD | —/fluorescence | 106) |
Somatostatin receptors (SSTRs) comprised of five subtypes (SSTR1–SSTR5) are transmembrane GPCRs37) and overexpressed in a variety of tumors such as gliomas, breast cancer, small cell lung cancer, and neuroendocrine tumors.38–40) Native somatostatin peptides derived from pre-pro-SST, SST-28, and SST-14 exhibit high affinity for SSTRs and are studied as attractive targeting agents for cancer therapy.41,42) However, the in vivo half-life of these peptides was very short due to enzymatic degradation. Therefore, somatostatin analogue peptides such as octreotide (a cyclic octapeptide) that could resist enzymatic degradation were developed and applied clinically because of their high inhibitory activity against hormones including growth hormone, prolactin, thyroid-stimulating hormone (TSH), insulin, and glucagon.41) In addition, several drug-conjugated or radiolabeled somatostatin analogues were developed using octreotide. As a drug-conjugated octreotide, octreotide-conjugated paclitaxel (PTX) was used to target SSTR2 selectively and internalized in SSTR-expressing MCF-7 human breast cancer cells, followed by apoptosis.43) [111In-DTPA0]-octreotide (Octreoscan) was developed as the first radiolabeled somatostatin analogue and has been used for scintigraphy imaging in primary and metastatic neuroendocrine tumors.44) The other octreotide chelator conjugates, 1,4,7,10-tetraazacyclodocecane-N,N′,N″,N‴-tetraacetic acid (DOTA)-d-Phe1-Tyr3-octreotide (DOTATOC) and DOTA-d-Phe1-Tyr3-octreotate (DOTAT ATE), showed high affinity to SSTR2.45) In addition, the positron emitter 68Ga-labeled DOTATOC was developed as a PET probe and showed clear images of neuroendocrine tumors in PET/computed tomography (CT).46) Octreotide-modified PEGylated liposome-encapsulated cantharidin was developed to decrease the systemic toxicity of cantharidin. It markedly inhibited the growth of MCF-7 cells and showed lower cytotoxic activity than free cantharidin.47) Zhang et al.48) synthesized octreotide–PEG–distearoylphosphatidylethanolamine (DSPE) and developed doxorubicin (DOX)-loaded octreotide-modified PEGylated liposomes to facilitate the intracellular delivery of DOX. Compared with the control liposome, the octreotide-modified PEGylated liposome exhibited high toxicity to SSTR2-positive cells via receptor-mediated endocytosis.48) Abou et al.49) developed 89Zr- and gadolinium (Gd)-labeled PEGylated liposomes modified with octreotide for PET/MRI dual-modality imaging. 89Zr is a positron emitter, and Gd is an MRI contrast medium. The 89Zr-Gd PEGylated liposomes modified with octreotide specifically recognized SSTR2 in vitro and in vivo, and PET/MRI exhibited clear images of SST2-expressing tumors using the liposome.49) In addition, it was reported that several SST peptide analogues such as BIM-23926 for SSTR1, BIM-23197 for SSTR2, BIM-23268 for SSTR5, BIM-23244 for SSTR2 and −5, and TT-232 for SSTR1 and −4 have antitumor effects.50–52) These SST peptide analogues are expected to become cancer-targeting ligands for DDS and molecular imaging.
2.2. Integrin-Targeted PeptidesIntegrins are transmembrane heterodimeric receptors that mediate cell adhesion in cell–cell and cell–matrix interactions.53) There are at least 24 integrin αβ heterodimers formed by 18α subunits and 8β subunits.53) Among them, integrin αvβ3, αvβ5, and α5β1 are upregulated in tumor cells and associated with angiogenesis, tumor growth, and metastasis.54) Therefore, peptide ligands for integrins are promising agents for drug delivery and molecular imaging.
The tripeptide Arg-Gly-Asp (RGD) was reported as a ligand for αvβ3 integrin overexpressed in solid tumors.55) The RGD-modified PEGylated liposome-encapsulated DOX enhanced drug accumulation in tumor cells by internalization through the integrin receptor-mediated endocytosis pathway and showed antitumor effects.56) To enhance the targeting efficacy, cyclic RGD-modified PEGylated liposomes were developed. Cyclic RGD peptides such as c(RGDfK), c(RGDfC), and RGD10 (DGARYCRGDCFDG) were more stable at neutral pH compared with the noncyclic RGD peptide and thus were able to resist proteolysis.36,57,58) In addition, they showed high affinity to αvβ3 integrin in human breast cancer Bcap-37, colon cancer HT29, and melanoma A375 cells.57,59) As imaging agents, RGD-modified probes were reported, such as [18F]Galacto-RGD, [18F]Alfatide, [68Ga]NOTA-PRGD2, 99mTc-HYNIC-3PEG4-E[c(RGDfK)2], and 64Cu-DOTA-QD-RGD. [18F]Galacto-RGD was the first RGD-modified PET tracer in clinical trials and allowed the visualization of tumors in vivo.60) Moreover, [18F]Galacto-RGD did not accumulate in the normal brain, unlike 18F-fluorodeoxyglucose (FDG), when used clinically as a PET tracer, suggesting that the RGD PET tracer can be applied to the imaging of glioma. [18F]Alfatide showed a higher tumor/background ratio in brain metastases compared with before the affinity was optimized.61) The metal complex of 68Ga and NOTA-PRGD2, which consists of a di-RGD peptide and the chelating moiety 1,4,7-triazacyclononane-N,N,N-triacetic acid (NOTA), was synthesized in a shorter time and under milder conditions compared with the 18F-labeling agent [18F]Galacto-RGD.62) Furthermore, the [68Ga]NOTA-PRGD2 showed clear images of lung lesions in PET/CT and was more advantageous than 18F-FDG in patients with lung cancer.62) The gamma emitter 99mTc-labeled HYNIC-3PEG4-E[c(RGDfK)2] was also synthesized as a SPECT/CT probe, shows high binding affinity to integrin αvβ3, and is used as a pretreatment screening tool in breast cancer.63,64) 64Cu-DOTA-QD-RGD is a dual-modality imaging probe, and the quantum dot (QD) surface was modified with about 90 RGD peptides and DOTA chelators for integrin αvβ3-targeted PET/near-infrared (NIR) fluorescence imaging. In the U87MG tumor model, 64Cu-DOTA-QD-RGD exhibited specific binding to integrin αvβ3 and provided sufficiently clear tumor images in both PET and optical NIR imaging.65)
ATN-161 (Ac-Pro-His-Ser-Cys-Asn-NH2) is a non-RGD peptide that specifically recognizes integrin α5β1, with potent antitumor and antimetastatic activity.66) The ATN-161-modified PEGylated DOX liposome was obtained by coupling the surface of PEGylated DOX liposomes and the ATN-161 lysine analogue. Confocal microscopy imaging showed that the cellular uptake of ATN-161-modified liposomes is mediated by integrin-mediated endocytosis. In both human umbilical vein endothelial cells (HUVECs) and breast cancer cells, ATN-161-modified PEGylated DOX liposomes showed significant antitumor effects.67)
2.3. Transferrin Receptor-Targeted PeptidesIron is an essential element for life, and intracellular iron is tightly regulated by iron homeostasis.68) Transferrin receptors (TFRs) 1 and 2 are transmembrane glycoproteins. The receptors regulate the cellular uptake of the complex of iron and transferrin (TF, an iron-binding protein) and are associated with the cellular process.69) Although the expression of TFR1 is low in many normal human tissues, it is increased in malignant cells including bladder-transitional cell carcinoma, breast cancer, glioma, lung adenocarcinoma, chronic lymphocytic leukemia, and non-Hodgkin’s lymphoma.70–74) In addition, it was reported that the expression level of TFR1 correlates with tumor stage and progression.75)
The blood–brain barrier (BBB) regulates the transport of substances including anticancer drugs through tight junction and P-glycoprotein.76) Because of the high expression of TFR in the BBB, TF-conjugated liposomes mainly cross the BBB via receptor-mediated endocytosis. Dual-targeting DOX liposomes conjugated with TF and folate showed antitumor effects in C6 glioma cells.77) Recently, photothermal and photodynamic therapy (PTT/PDT) has been a focus of interest because of the anticancer effect produced by heat and singlet oxygen (1O2). In order to apply PTT/PDT therapy and molecular imaging, IR780 (tricarbocyanine NIR dye)-loaded TF nanoparticles were developed. The nanoparticles showed a high tumor/background ratio in CT26 colorectal cancer cell-bearing mice and significantly inhibited tumor growth.78) However, there are some doubts about those nanoparticles in terms of safety, production cost, and modification as a drug carrier since TF is a full-length protein.
Peptide T7 (HAIYPRH) was discovered using a phage-display method and showed higher TFR binding activity compared with TF.79) TFR-targeted liposomes modified with T7-loaded PTX exhibited antitumor activity against A2780 human ovarian cancer cells.80) To increase the cellular uptake, CPPs such as TAT peptide were conjugated to the surface of T7-modified liposomes81) and exhibited high efficacy through receptor/transporter-dependent and -independent pathways.82) The PDT probe TPETH-2T7, a red-emissive aggregation-induced emission tetraphenylethenethiophene (TPETH) conjugated with T7 showed a fluorescence turn-on response in real-time imaging, and the PDT effect was superior when TPETH-2T7 was localized at the cell membrane of MDA-MB-231 breast cancer cells.83) Recently, a hybrid peptide (THRPPMWSPVWPGGGKLLLKLLKKLLKLLKKK) including a TF receptor-binding peptide and lytic peptide has been designed and shown to have cytotoxic activity and to inhibit tumor progression.84)
2.4. HER2-Targeted PeptidesHER2 belongs to the EGF family of type 1 tyrosine kinase and is overexpressed in many cancers such as breast, ovarian, endometrial, gastric, pancreatic, and prostate cancers.85) In particular, HER2 is overexpressed in around 20–30% of breast cancers, and HER2 status is considered to be a marker of poor prognosis.86)
As a HER2-targeted drug, trastuzumab, a recombinant monoclonal antibody that binds to the extracellular region of HER2, was developed and showed efficacy in metastatic HER2-positive breast cancer patients.87) In addition, higher therapeutic effects are achieved when trastuzumab is combined other chemotherapy.88) Generally, immunonanoparticles are expected to increase tumor selectivity compared with unmodified anticancer agents. Therefore, anti-HER2 immunoliposome-functionalized trastuzumab was developed, and anti-HER2 immunoliposome-encapsulated anticancer agents such as DOX or PTX showed potent antitumor effects in HER2-overexpressing breast cancer cells.89) However, recombinant monoclonal antibodies are relatively costly due to their complicated production procedures including expression and purification. Recently, single-chain Fv fragments of trastuzumab have been applied to immunoliposomes, although the problems of cost and antigenicity remain.
On the other hand, peptide ligands can be produced cost-effectively and have low antigenicity. Therefore, HER2-specific peptide ligands have attracted attention recently. Karasseva et al.90) discovered the peptide KCCYSL using the phage-display method and reported its binding activities to HER2-positive human breast and prostate carcinoma cells. Bandekar et al.91) reported a pH-responsive PEGylated DOX liposome modified with KCCYSL. This liposome specifically binds to and internalizes in HER2-positive cells, and subsequently pH-tunable vesicles release DOX rapidly and extensively. In HER2-overexpressing BT474 breast cancer cell-bearing nude mice, this liposome inhibited tumor growth.91) The short peptide LTVSPWY was also identified as a HER2-binding peptide.92) As an MRI imaging probe, PEGylated chitosan-modified LTVSPWY (LTVSPWY-PEG-CS) was prepared using the solvent-diffusion method, and LTVSPWY-PEG-CS identified tumors rapidly and efficiently in in vivo experiments.93) Moreover, the peptide AHNP (FCDGFYACYADVGGG) was designed from a heavy-chain CDR3 loop of trastuzumab, and its HER2-specific affinity was reported.94) Zahmatkeshana et al.95) synthesized AHNP–PEG–DSPE with three glycine amino acids as a spacer and applied it to AHNP-modified PEGylated DOX liposomes. In HER2-positive TUBO tumor-bearing mice, this liposome exhibited marked inhibition of tumor growth.95)
2.5. OthersAs another target for chemotherapy, LHRH receptors are overexpressed in several cancers such as endometrial, ovarian, prostate, breast, bladder, colorectal, and pancreatic cancers.96,97) Based on the native LHRH peptide, short LHRH peptide analogues were developed as tumor-specific ligands.98,99) LHRH peptide analogues linked to anticancer agents via ester bonds were also synthesized and improved the accumulation of the linked agents. As an example, a clinical trial was conducted of the peptide [D-Lys6]LHRH linked to DOX (AEZS-108).100) However, the binding affinity of the peptide analogue may decrease because the anticancer drug directly conjugated to the peptide analogue interferes with the binding function of the peptide analogue. LHRH receptor-targeting nanoparticles were then developed to overcome that drawback, and, for example, liposomes composed of DSPE–PEG, DSPE–PEG–Cy5.5, DSPE–PEG–LHRH peptide, and PTX showed efficacy in cancer treatment and imaging.101)
Tripeptide Asn-Gly-Arg (NGR) is a ligand of aminopeptidase N (APN/CD13) that is overexpressed in tumor cells and angiogenic blood vessels.102) NGR-modified PEGylated liposomes efficiently delivered c-myc siRNA to the cytoplasm of HT1080 fibrosarcoma cells after intravenous injection. As a result, c-myc suppression and evoked cellular apoptosis were observed in the tumor.103) In addition, NGR-modified PEG-b-PLA polymeric micelles loaded with docetaxel was quantitatively accumulated in HT1080 fibrosarcoma cells and HUVECs and showed antitumor activity.104) A combination of NGR, thermosensitive liposomes, and DOX conjugated with CPP was developed and showed significant tumor growth inhibition in HT1080 fibrosarcoma cells.105) In addition, NGR peptide conjugated to imaging agents including fluorescent dyes, QDs, micelles, and liposomes has the potential to visualize tumors. PEGylated CdSe/AnS QDs modified with NGR peptide specifically recognized CD31 and clearly showed glioma-associated vessels in a fluorescent imaging system.106)
To date, several peptide ligands have been developed and incorporated into nanoparticles for DDS and molecular imaging. Peptide-modified PEGylated liposome-encapsulated anticancer drugs have attracted particular attention in cancer-targeted therapy, and these liposomes combined with imaging agents allow the visualization of tumors or their microenvironments.
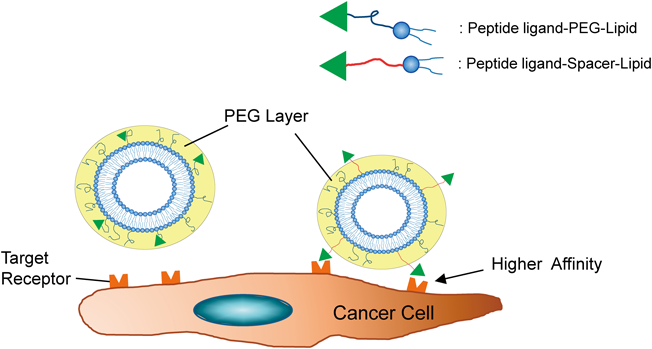
PEG on the surface of liposomes was reported to take mushroom or brush conformations and construct a PEG layer,107) and PEG is known to confer steric stabilization effects on liposomes for passive targeting. In addition, PEG has been utilized as a spacer, and peptide ligands are conjugated to the end of PEG lipids. However, in PEGylated liposomes modified with conjugated ligand–lipid, sufficient effects of ligand-mediated active targeting are not expected due to the embedding in the PEG layer. Therefore, it is important to display peptide ligands efficiently outside the PEG layer (Fig. 1). To overcome this drawback, Stefanick et al.108) used a Glu-Gly repeat sequence (EG)n instead of PEG spacers and examined the cellular uptake of PEGylated liposomes modified with a HER2- or VLA-4-antagonistic peptide–EG linker. Compared with PEGylated liposomes using PEG spacers, the combination of liposomal PEG350 and EG12 spacer dramatically improved its accumulation by 9- and 100-fold in breast cancer and multiple myeloma cells, respectively.108) As another linker, Veneti et al.109) reported an elastin-like peptide (ELP) linker for RGD-targeted liposomes. Since the ELP linker showed conformational change in an acidic tumor microenvironment, the liposome-modified RGD-ELP linker significantly recognized both MDA-MB-231 and HCC1806 breast cancer cells.109) These findings indicate that spacer peptides contribute to the efficacy of peptide–ligand-mediated active targeting.
The combination of nanoparticle-modified peptide ligands and several stimuli such as pH, temperature, enzymes, and ultrasound is a promising strategy for active targeting in cancer treatment or molecular imaging.110,111) The pH-sensitive peptide GALA occurs as a random coil conformation at pH 7, whereas it converted to an amphipathic α-helix structure at pH 5.112) This conformational change of GALA assisted the endosomal escape of PEGylated liposomes to the cytosol. Hyperthermia and photothermia by magnetic or optical nanoparticles increased not only the tumor temperature but also the tumor vascular permeability of nanoparticles in solid tumors.113,114) Extracellular enzyme-catalyzed dePEGylation is effective for drug delivery because endosomal escape inhibited by PEGylation was interrupted. Hatakeyama et al.115) synthesized PEG–peptide–DOPE including a peptide sequence cleaved by matrix metalloproteinase-2 and prepared a GALA-modified envelope-type nanodevice containing PEG–peptide–DOPE. This nanoparticle enhanced endosomal escape and subsequently promoted siRNA delivery both in vitro and in a tumor-bearing mouse model.115) Ultrasound (US) is a common medical diagnostic imaging modality. Sonoporation induced by US and bubble liposomes modified with AG73 peptide for syndecans or bubble lipoplex-modified mannose created temporal pores on the cell membrane, and anticancer drugs, siRNA, and plasmid DNA were delivered to tagged tumors or organs.116–120) Thus, internal and external stimuli are effective tools to improve drug targeting.
In summary, peptides binding to cancer-specific receptors have great potential for drug targeting, and peptide-modified nanoparticles are expected to be used clinically for DDS and molecular imaging.
The authors declare no conflict of interest.