Regular Articles
Synthesis, Molecular Docking and Biological Evaluation of Quinolone Derivatives as Novel Anticancer Agents
2018 Volume 66 Issue 1 Pages 55-60

Details
2018 Volume 66 Issue 1 Pages 55-60
A series of novel quinolone derivatives (8a–j) were synthesized, and their anticancer activities were tested in human cancer cell lines, human lung carcinoma cell (A549), human promyelocytic leukemia cell (HL-60), and human cervical cancer cell (Hela). Compound 8i was found to be 5-times more potent in cell-killing activity for cell lines A549, HL-60, and Hela than the positive control irinotecan or cisplatin, with IC50 of 0.009, 0.008 and 0.010 µM, respectively. The docking study revealed that compound 8i might have strong interactions with the active site of DNA-topoisomerase I.
Quinolone as a privileged scaffold represents one of the most important structural unit prevalent in various naturally occurring and bioactive compounds.1) Quinolones consist of a bicyclic ring structure (Fig. 1) in which there is a substitution at position N-1, with various moieties. Most of the current agents have a carboxyl group at position 3, a keto group at position 4, a fluorine atom at position 6 and a nitrogen heterocycle at position 7.2,3) In the late 1980 s, reports emerged describing experimental antibacterial quinolones having significant potency against eukaryotic topoisomerases (Top) and showing cytotoxic activity against tumor cell lines.4–7)

Li and colleagues designed and synthesized a series of quinolone derivatives as potential Top I inhibitors for cancer treatment.8–10) Rajulu et al. designed a series of fluoroquinolones displaying good growth inhibition activities against human lung carcinoma cell (A549) and colon carcinoma (HCT-116).11) Recently, our group discovered a novel series of Top I inhibitors with quinolone scaffold. Quinolone derivative 1 was the most potent compound we synthesized.12) Several important series of antimicrobial agents are associated with particular N-1 substituents. Tosufloxacin (2) and difloxacin (3) (Fig. 2) with the substituent of fluorine atom in the aromatic ring enhanced both potency against Gram-positives and pharmacokinetics.13) The antibacterial tosufloxacin (2) can be viewed as intermediate agents on the evolutionary path toward both antibacterial and anticancer quinolone derivatives.7) Using a scaffold modification strategy, our team changed the N-1 cyclopropyl group into N-1 aryl substituents with fluorine in the aromatic ring (Fig. 2).

The method for the preparation of novel N-fluoroaromatic substituted piperazinylquinolone derivatives 8a–j relies on the Grohe–Heitzer cycloacylation reaction.14,15) The synthetic route is outlined in Chart 1. The commercially available compound 4 was subjected to an addition–elimination reaction with a substituted primary amine, and the obtained product 5 was cyclised in a tandem addition–elimination reaction at the ortho position. Compound 7 was obtained by displacing the chlorine atom in compound 6 with anhydrous piperazine. Then the target compound 8 was prepared via a two-step one-pot tandem process.12)

All the target compounds (8a–j) were evaluated for their in vitro cytotoxic activity against three different human cancer cell lines, A549, human promyelocytic leukemia cell (HL-60) and human cervical cancer cell (Hela) by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay with irinotecan and cisplatin as positive control compounds. The assessments of anticancer activities were expressed as the concentration inhibiting 50% of cancer cell growth (IC50). The Top I inhibitory activity assays was carried out using a topoisomerase I drug screening kit. The results are summarized in Table 1 and Fig. 3. As illustrated in Table 1, these compounds exhibited significant antitumor activity. Among them, compound 8i displayed the most potent inhibitory activity (IC50=0.009 µM for A549, IC50=0.008 µM for HL-60, and IC50=0.010 µM for Hela), which was better than the positive control irinotecan (IC50=0.032 µM for A549, IC50=0.044 µM for HL-60, and IC50=0.038 µM for Hela) and cisplatin (IC50=0.048 µM for A549, IC50=0.057 µM for HL-60, and IC50=0.047 µM for Hela). As expected, compound 8i showed excellent Top I inhibitory activity comparable to irinotecan and cisplatin.
Subsequently, structure–activity relationships (SAR) studies were inferred from Table 1. In general, target compounds with N-1 substituent of 2,4-difluorophenyl group (8f–j) showed more potent activities than those with N-1 substituent of 4-fluorophenyl group (8a–e). In three different human cancer cell lines, using electron withdrawing group (Cl, CF3) in the phenyl moiety as in compounds 8b–e and 8g–j increase the reactivity rather than using electron donating group (CH3) in the phenyl moiety as in compounds 8a and 8f. And compounds 8i and 8j with trifluoromethyl in the phenyl moiety showed higher cytotoxic activity than in compounds 8g and 8h with chlorine atom in the phenyl moiety. It is noted that the substitution R2 at the 2-position of phenyl (8i) exhibiting better biological activities than the substitution R2 at the 3-position of phenyl (8j), which may be due to steric-hindrance effect.
| Compd. | IC50 (µM) | ||
|---|---|---|---|
| A549 | HL-60 | Hela | |
| 1 | 0.071 | 0.043 | 0.032 |
| 4 | >0.157 | >0.157 | >0.157 |
| 8a | >0.086 | 0.081 | >0.086 |
| 8b | >0.053 | 0.049 | 0.040 |
| 8c | 0.041 | 0.039 | 0.043 |
| 8d | 0.032 | 0.048 | 0.049 |
| 8e | 0.036 | 0.016 | 0.037 |
| 8f | >0.082 | 0.050 | 0.069 |
| 8g | 0.039 | 0.044 | 0.045 |
| 8h | 0.038 | 0.033 | 0.029 |
| 8i | 0.009 | 0.008 | 0.010 |
| 8j | 0.028 | 0.018 | 0.010 |
| Irinotecan | 0.032 | 0.044 | 0.038 |
| Cisplatin | 0.048 | 0.057 | 0.047 |
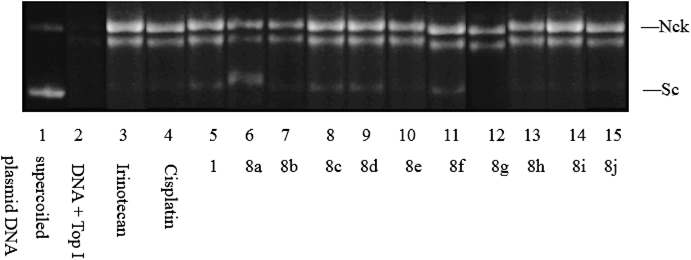
Inhibition of Top I at 1 µM. Lane 1: supercoiled plasmid DNA (pBR322); Lane 2: DNA+Top I; Lane 3: DNA+Top I+Irinotecan; Lane 4: DNA+Top I+Cisplatin; Lane 5–15: DNA+Top I+compounds (1, 8a, 8b, 8c, 8d, 8e, 8f, 8g, 8h, 8i and 8j). Sc—supercoiled DNA, Nck—nicked open circular DNA.
Docking was performed against DNA-Top I because it is reported as possible anticancer target of quinolone derivatives.16–19) In order to understand the binding conformation of the most potent cytotoxic activity of the compound 8i, its flexible molecular docking was carried out into the Top I (PDB code: 1K4T) active site using CDOCKER protocol of Discovery Studio 2.1.
The binding modes of compound 8i and Top I-DNA complex were depicted in Fig. 4. The amino acid residue which had interaction with Top I-DNA complex were labeled in Figs. 4A and 4B. In the binding mode, compound 8i was nicely bound to the Top I-DNA complex active site via six hydrogen bond, two cation–π interaction and ten π–π interaction. The nitrogen atom of guanidine group and one of the sulfonyl oxygen formed two hydrogen bond with amino of LYS 751. The fluorine atom at the 2-position of phenyl and the carboxyl group of 8i formed two hydrogen bond with amino of ARG 364 and two hydrogen bond with base pair of TGP 11, respectively. And the benzene ring with two fluorine atom associated with LYS 532 and ARG 364 by two cation–π interactions. The quinolone skeleton and the benzene ring of compound 8i stabilized by base-stacking interactions with both the upstream (−1) and downstream (+1) base pairs with ten π–π interactions.
The enzyme surface model was shown in Fig. 4C, which revealed that the molecule was well embedded in the active pocket. This molecular docking results and the biological assay data suggested that compound 8i was a potential Top I inhibitors as anti-cancer agents.

The dotted lines show the hydrogen bonds and the solid lines show the π–cation and π–π interactions.


The protein is represented by molecular surface. Compound 8i is depicted by balls.
We have designed and synthesized a novel series of quinolone derivatives (8a–j). These compounds exhibited in vitro cytotoxic activity against A549, HL-60, and Hela cells. Docking simulations were performed to position most active compound 8i into the Top I-DNA complex active site to determine the probable binding conformation and the results confirmed that the compound was a potential Top I inhibitor.
Melting points (m.p.) were determined on Büchi B-540 melting point apparatus and are uncorrected. 1H-NMR (300 MHz) spectra were recorded on a Bruker AV 300 MHz spectrometer. Mass spectra were obtained on a Thermo Finnigan LCQ-Advantage spectrometer (electrospray ionization (ESI), atmospheric pressure chemical ionization (APCI)), and high resolution (HR)-MS were carried out on an APEX (Bruker) mass III spectrometer. The compounds were dissolved in dimethyl sulfoxide (DMSO)-d6. Chemical shifts were reported in ppm (δ) relative to tetramethylsilane (TMS) (δ=0). Coupling constants, J, are reported in Hz, multiplicities being marked as: singlet (s), broad singlet (br s), doublet (d), triplet (t), quartet (q), multiplet (m).
General Procedure for Synthesis of Compounds(5a, b) Firstly, a solution of commercially available compound 4 (8g, 25 mmol) in 25 mL ethanol, was added with the solution of fluoro-substituted aniline (26.25 mmol) in 10 mL ethanol dropwise during 10 min. The reaction mixture was then stirred at 45°C for 6 h. The cooled reaction was filtered to give the desired product 5 as light yellow solid.
General Procedure for Synthesis of Compounds(6a, b) A solution of 5 (10 mmol), K2CO3 (2.2 g, 16 mmol) in 20 mL N,N-dimethylformamide (DMF) was stirred at 140°C for 2 h. The hot mixture was filtered rapidly, then the solution was allowed to stir at room temperature for 10 min until a yellow precipitate is formed. The solid was filtered, washed with water twice, and dried in vacuo.
General Procedure for Synthesis of Compounds(7a, b) The compound 6 (3.5 mmol), anhydrous piperazine (1.5 g, 17.5 mmol) and isopropanol (10 mL) were mixed and stirred at 130°C for 3 h, then the solvent was removed under reduced pressure. The residue was added with 10% NaOH (6 mL), and stirred at 90°C for 1.5 h, then added with activated carbon and stirred at reflux for 1 h, respectively. The hot mixture was filtered and the aqueous phase was then acidified with hydrochloric acid to pH 7. The mixture was stirred at room temperature until a precipitated is formed. The solid was filtered on a Buchner funnel to give the desired product 7.
General Procedure for Synthesis of Compounds(8a, j) Benzenesulfonyl chloride (1 mmol) in butanone (5 mL) was heated with stirring to 40°C, and cyanamide solution (50%) was added dropwise, then the temperature was raised to 60°C and stirring continued for 3 h. The compound 7 (0. 8 mmol) was added and heated to 80°C for 3 h. After cooling to 40°C, the reaction mixture was poured into cold water while stirring, white crystals or powders were precipitated, filtered, washed with water, and dried. Analytically pure samples were obtained by recrystallization from aqueous ethanol.
(Z)-Methyl 2-(2,4-Dichloro-5-fluorobenzoyl)-3-((4-fluorophenyl)amino)acrylate (5a)1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 3.80 (s, 3H, –CH3), 6.74–6.94 (m, 4H, –C6H4), 7.55 (d, 1H, J=4.0 Hz, 6-H), 7.81 (d, 1H, J=4.0 Hz, 3-H), 9.79 (s, 1H, –NH). MS (ESI): 385.0.
(Z)-Methyl 2-(2,4-Dichloro-5-fluorobenzoyl)-3-((2,4-difluorophenyl)amino)acrylate (5b)1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 3.83 (s, 3H, –CH3), 6.60–6.78 (m, 3H, –C6H3), 7.61 (d, 1H, J=4.0 Hz, 6-H), 7.83 (d, 1H, J=4.0 Hz, 3-H), 9.56 (s, 1H, –NH). MS (ESI): 403.0.
Methyl 7-Chloro-6-fluoro-1-(4-fluorophenyl)-4-oxo-1,4-dihydroquinoline-3-carboxylate (6a)1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 3.82 (s, 3H, –CH3), 7.09–7.18 (m, 4H, –C6H4), 7.25 (d, 1H, J=4.0 Hz, 8-H), 7.66 (d, 1H, J=4.0 Hz, 5-H), 8.29 (s, 1H, 2-H). MS (ESI): 349.0.
Methyl 7-Chloro-1-(2,4-difluorophenyl)-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylate (6b)1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 3.82 (s, 3H, –CH3), 6.82–7.12 (m, 3H, –C6H3), 7.18 (d, 1H, J=4.0 Hz, 8-H), 7.66 (d, 1H, J=4.0 Hz, 5-H), 8.25 (s, 1H, 2-H). MS (ESI): 367.0.
6-Fluoro-1-(4-fluorophenyl)-4-oxo-7-(piperazin-1-yl)-1,4-dihydroquinoline-3-carboxylic Acid (7a)1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 1.20 (s, 1H, –NH), 2.82–2.85 (m, 4H, –CH2, piperazine), 3.53–3.63 (m, 4H, –CH2, piperazine), 6.48 (d, 1H, J=4.0 Hz, 8-H), 7.07–7.19 (m, 4H, –C6H4), 7.87 (d, 1H, J=4.0 Hz, 5-H), 8.32 (s, 1H, 2-H). MS (ESI): 385.1.
1-(2,4-Difluorophenyl)-6-fluoro-4-oxo-7-(piperazin-1-yl)-1,4-dihydroquinoline-3-carboxylic Acid (7b)1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 1.22 (s, 1H, –NH), 2.81–2.85 (m, 4H, –CH2, piperazine), 3.34–3.70 (m, 4H, –CH2, piperazine), 6.75 (d, 1H, J=4.0 Hz, 8-H), 6.85–7.13 (m, 4H, –C6H4), 8.02 (d, 1H, J=4.0 Hz, 5-H), 8.20 (s, 1H, 2-H). MS (ESI): 403.1.
6-Fluoro-1-(4-fluorophenyl)-4-oxo-7-(4-(N-tosylcarbamimidoyl)piperazin-1-yl)-1,4-dihydroquinoline-3-carboxylic Acid (8a)M.p. 200–202°C, Yield 61%, 1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 2.45 (s, 3H, –CH3), 3.38–3.59 (m, 4H, –CH2, piperazine), 3.78–3.88 (m, 4H, –CH2, piperazine), 6.98 (d, 1H, J=4.0 Hz, 8-H), 7.08–7.10 (m, 4H, –C6H4), 7.30–7.65 (m, 4H, –C6H4), 8.01 (d, 1H, J=4.0 Hz, 5-H), 8.20 (s, 1H, 2-H); HR-MS Calcd for C28H25F2N5O5S [M−]: 581.1544. Found: 581.1541.
7-(4-(N-((2-Chlorophenyl)sulfonyl)carbamimidoyl)piperazin-1-yl)-6-fluoro-1-(4-fluorophenyl)-4-oxo-1,4-dihydroquinoline-3-carboxylic Acid (8b)M.p. 216–218°C, Yield 73%, 1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 3.35–3.52 (m, 4H, –CH2, piperazine), 3.75–3.81 (m, 4H, –CH2, piperazine), 6.91 (d, 1H, J=4.0 Hz, 8-H), 7.06–7.7.08 (m, 4H, –C6H4), 7.40–7.78 (m, 4H, –C6H4), 7.92 (d, 1H, J=4.0 Hz, 5-H), 8.43 (s, 1H, 2-H); HR-MS Calcd for C27H22ClF2N5O5S [M−]: 601.0998. Found: 601.0998.
7-(4-(N-((2,6-Dichlorophenyl)sulfonyl)carbamimidoyl)piperazin-1-yl)-6-fluoro-1-(4-fluorophenyl)-4-oxo-1,4-dihydroquinoline-3-carboxylic Acid (8c)M.p. 211–213°C, Yield 71%, 1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 3.40–3.54 (m, 4H, –CH2, piperazine), 3.76–3.84 (m, 4H, –CH2, piperazine), 6.83 (d, 1H, J=4.0 Hz, 8-H), 7.10–7.13 (m, 4H, –C6H4), 7.49–7.73 (m, 3H, –C6H3), 8.01 (d, 1H, J=4.0 Hz, 5-H), 8.26 (s, 1H, 2-H); HR-MS Calcd for C27H21Cl2F2N5O5S [M−]: 635.0609. Found: 635.0601.
6-Fluoro-1-(4-fluorophenyl)-4-oxo-7-(4-(N-((2-(trifluoromethyl)phenyl)sulfonyl)carbamimidoyl)piperazin-1-yl)-1,4-dihydroquinoline-3-carboxylic Acid (8d)M.p. 207–209°C, Yield 60%, 1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 3.35–3.57 (m, 4H, –CH2, piperazine), 3.75–3.84 (m, 4H, –CH2, piperazine), 7.01 (d, 1H, J=4.0 Hz, 8-H), 7.08–7.09 (m, 4H, –C6H4), 7.54–7.75 (m, 4H, –C6H4), 8.01 (d, 1H, J=4.0 Hz, 5-H), 8.21 (s, 1H, 2-H); HR-MS Calcd for C28H22F5N5O5S [M−]: 635.1262. Found: 635.1260.
6-Fluoro-1-(4-fluorophenyl)-4-oxo-7-(4-(N-((3-(trifluoromethyl)phenyl)sulfonyl)carbamimidoyl)piperazin-1-yl)-1,4-dihydroquinoline-3-carboxylic Acid (8e)M.p. 205–206°C, Yield 65%, 1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 3.44–3.54 (m, 4H, –CH2, piperazine), 3.76–3.84 (m, 4H, –CH2, piperazine), 6.99 (d, 1H, J=4.0 Hz, 8-H), 7.10–7.14 (m, 4H, –C6H4), 7.53–7.92 (m, 3H, –C6H4), 7.93 (d, 1H, J=4.0 Hz, 5-H), 8.19 (s, 1H, –C6H4), 8.37 (s, 1H, 2-H); HR-MS Calcd for C28H22F5N5O5S [M−]: 635.1262. Found: 635.1259.
1-(2,4-Difluorophenyl)-6-fluoro-4-oxo-7-(4-(N-tosylcarbamimidoyl)piperazin-1-yl)-1,4-dihydroquinoline-3-carboxylic Acid (8f)M.p. 203–204°C, Yield 62%, 1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 2.28 (s, 3H, –CH3), 3.37–3.58 (m, 4H, –CH2, piperazine), 3.78–3.87 (m, 4H, –CH2, piperazine), 7.01 (d, 1H, J=4.0 Hz, 8-H), 7.09–7.42 (m, 3H, –C6H3), 7.44–7.92 (m, 4H, –C6H4), 8.27 (d, 1H, J=4.0 Hz, 5-H), 8.93 (s, 1H, 2-H); HR-MS Calcd for C28H24F3N5O5S [M−]: 599.1450. Found: 599.1449.
7-(4-(N-((2-Chlorophenyl)sulfonyl)carbamimidoyl)piperazin-1-yl)-1-(2,4-difluorophenyl)-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic Acid (8g)M.p. 215–218°C, Yield 72%, 1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 3.36–3.58 (m, 4H, –CH2, piperazine), 3.79–3.86 (m, 4H, –CH2, piperazine), 7.06 (d, 1H, J=4.0 Hz, 8-H), 7.18–7.44 (m, 3H, –C6H3), 7.52–8.03 (m, 4H, –C6H4), 8.26 (d, 1H, J=4.0 Hz, 5-H), 8.95 (s, 1H, 2-H); HR-MS Calcd for C27H21ClF3N5O5S [M−]: 619.0904. Found: 619.0902.
7-(4-(N-((2,6-Dichlorophenyl)sulfonyl)carbamimidoyl)piperazin-1-yl)-1-(2,4-difluorophenyl)-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic Acid (8h)M.p. 210–212°C, Yield 79%, 1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 3.47–3.52 (m, 4H, –CH2, piperazine), 3.79–3.84 (m, 4H, –CH2, piperazine), 7.06 (d, 1H, J=4.0 Hz, 8-H), 7.18–7.26 (m, 3H, –C6H3), 7.48–7.72 (m, 3H, –C6H3), 8.28 (d, 1H, J=4.0 Hz, 5-H), 8.95 (s, 1H, 2-H); HR-MS Calcd for C27H20Cl2F3N5O5S [M−]: 653.0514. Found: 653.0519.
1-(2,4-Difluorophenyl)-6-fluoro-4-oxo-7-(4-(N-((2-(trifluoromethyl)phenyl)sulfonyl)carbamimidoyl)piperazin-1-yl)-1,4-dihydroquinoline-3-carboxylic Acid (8i)M.p. 205–207°C, Yield 68%, 1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 3.36–3.58 (m, 4H, –CH2, piperazine), 3.74–3.83 (m, 4H, –CH2, piperazine), 6.84 (d, 1H, J=4.0 Hz, 8-H), 7.08–7.22 (m, 3H, –C6H3), 7.53–7.75 (m, 4H, –C6H4), 8.25 (d, 1H, J=4.0 Hz, 5-H), 8.67 (s, 1H, 2-H); HR-MS Calcd for C28H21F6N5O5S [M−]: 653.1168. Found: 653.1163.
1-(2,4-Difluorophenyl)-6-fluoro-4-oxo-7-(4-(N-((3-(trifluoromethyl)phenyl)sulfonyl)carbamimidoyl)piperazin-1-yl)-1,4-dihydroquinoline-3-carboxylic Acid (8j)M.p. 202–204°C, Yield 69%, 1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 3.40–3.55 (m, 4H, –CH2, piperazine), 3.75–3.83 (m, 4H, –CH2, piperazine), 6.82 (d, 1H, J=4.0 Hz, 8-H), 7.08–7.23 (m, 3H, –C6H3), 7.53–7.92 (m, 3H, –C6H4), 8.24 (d, 1H, J=4.0 Hz, 5-H), 8.28 (s, 1H, –C6H4), 8.67 (s, 1H, 2-H); HR-MS Calcd for C28H21F6N5O5S [M−]: 653.1168. Found: 653.1161.
Anti-proliferation AssayThe antiproliferative activities of the prepared compounds against A549, HL-60 and Hela cells were evaluated using a standard MTT-based colorimetric assay. Target tumor cell lines were grown to log phase in RPMI 1640 medium supplemented with 10% fetal bovine serum. After diluting to 1×106 cells mL−1 with the complete medium, 100 µL of the obtained cell suspension was added to each well of 96-well culture plates. The subsequent incubation was permitted at 37°C, 5% CO2 atmosphere for 24 h before the cytotoxicity assessments. Each concentration was in triplicate, and Irinotecan was used as the positive control. After 72 h incubation at 37°C, 5% CO2 atmosphere, 10 µL of MTT solution in Dulbecco’s modified Eagle’s medium (DMEM) (Invitrogen, U.S.A.) was added to each well. After three hours incubation at 37°C, 150 µL DMSO was added to each well. The plates were then vibrated for 10 min for complete dissolution. The optical absorbance was measured at 570 nm on an automated microplate spectrophotometer (Bio-Rad, U.S.A.). In all experiments three replicate wells were used for each drug concentration. The IC50 value was defined as the concentration at which 50% of the cells could survive. The results were summarized in Table 1.
Top I InhibitionTop I inhibition was assayed by determining relaxation of supercoiled DNA pBR322. The test compounds were dissolved in DMSO and diluted with the final concentration 1 or 10 µM respectively, the same method to irinotecan and cisplatin at the concentration of 1 or 10 µM. A mixture of 0.5 µg of plasmid pBR322 and 10 U top I was incubated with the test compounds in final volume of 20 µL (in DMSO) at 37°C for 30 min in relaxation buffer (20 mmol/L Tris–HCl (pH 7.8), 50 mmol/L KCl, 10 mmol/L MgCl2, 1 mmol/L dithiothreitol (DTT), 0.2 mmol/L ethylenediaminetetraacetic acid (EDTA)). The reactions were terminated by adding 2.5 µL of stop solution containing 10% sodium dodecyl sulfate (SDS), 0.2% bromophenol blue, 0.2% xylene cyanol and 30% glycerol. DNA samples were then electrophoresed on 1% agarose gel for 10 h with Tris-borate–EDTA running buffer. Gels were stained for 30 min in an aqueous solution of ethidium bromide and visualized by transillumination with UV light.
Molecular DockingThe pdb file about the crystal structure of DNA-Top I bound to Topotecan (PDB code: 1T8I)20) was obtained from the RCSB Protein Data Bank (http://www.pdb.org). The molecular docking procedure was performed by using CDOCKER protocol for receptor–ligand interactions of Discovery Studio 2.1. For ligand preparation, the 3D structures of 8i were generated and minimized using Discovery Studio 2.1. For protein preparation, the hydrogen atoms were added. The whole DNA-Top I domain defined as a receptor and the site sphere was selected based on the ligand binding location of Topotecan, then the Topotecan removed and the prepared ligand was placed during the molecular docking procedure. CHARMm was selected as the force field. The molecular docking was performed with a simulated annealing method.
We are grateful to the National Natural Science Foundation of China (No. 21306104, 21476128) and the Foundation of Educational Committee of Zhejiang Province of China (No. Y201329284) for financial support.
The authors declare no conflict of interest.