Review
Creation of Novel Toxic Gas Surrogates and the Development of Safe and Facile Catalytic Reactions
2018 Volume 66 Issue 1 Pages 1-19
Details
2018 Volume 66 Issue 1 Pages 1-19
The use of toxic gas surrogates in organic reactions instead of the gas itself contributes to enhancing the safety, practicality, and efficiency of the reactions involved. Our efforts toward the creation of toxic gas surrogates and the development of a series of catalytic reactions using these surrogates are described. Improvements in substrate scope during the hydroesterification of alkenes using formates facilitated by the Ru–imidazole catalyst system provided the opportunity to discover that phenyl formate is a useful carbon monoxide (CO) surrogate for the generation of CO and phenol under weakly basic conditions. This discovery triggered the development of highly reactive but stable CO surrogates and a variety of Pd-catalyzed carbonylative transformations. N-Formylsaccharin facilitated the use of additional nucleophiles in carbonylation reactions that provided access to a variety of carbonyl compounds. Detailed experimental and theoretical mechanistic studies into the generation of CO from phenyl formate suggest that CO generation proceeds via a concerted E2 α-elimination. Furthermore, a known surrogate of sulfur dioxide was applied for the first time to the selective syntheses of cyclic sulfonamides and sulfinamides, confirming that the surrogate operates as an “S=O” source. Notably, the reactions described herein are scalable and can be performed without the use of external toxic gases and specialized reaction vessels; they are easy and simple to perform and demonstrate enormous potential for industrial application.
Most gaseous compounds generally require special handling techniques for transportation, storage, and use in organic reactions. However, some are regarded as important chemical feedstocks that are widely applied to the industrial production of a variety of compounds due to their simple structures, low molecular weights, and ready availabilities at low prices.1) For example, carbon monoxide (CO), located at the center of C1 chemistry,2) is naturally abundant and used in transition metal-catalyzed carbonylations to introduce carbonyl groups into various organic molecules.3–7) Sulfur dioxide (SO2) is mostly obtained by the oxidation of elemental sulfur collected from desulfurization processes in petrochemical plants,8,9) and is used for the synthesis of industrially important sulfuric acid and its derivatives.10)
There are many points to consider and special equipment to be prepared in order to conduct experiments involving gases; they must be carried out in appropriate environments that ensure the safety of users while complying with relevant regulations. Special laboratory equipment, such as pressure-resistant vessels and gas sensors, is also required. Furthermore, many high-pressure experiments necessitate complicated experimental manipulation. The most serious issue concerning the use of gas is toxicity. Since most gases used in organic synthesis are toxic for humans even at low concentrations, their exposure and leakage must be strictly suppressed during the reaction to ensure user safety. Therefore, difficulties associated with the handling of toxic gases that have unfavorable chemical and physical properties have significantly hampered their utilization in organic synthesis, especially on the small scale, rendering many research areas involving toxic gases largely unexplored.
Over the past few decades, the use of toxic gas “surrogates” has gained significant attention in the chemistry community.11–21) These surrogates are stable non-gaseous compounds that generate toxic gases through chemical reactions or physical stimuli. The resulting toxic gas is generated in a closed reaction vessel and is then used in organic reactions in the same manner as that stored in a cylinder, preventing direct contact with the user. Notably, the amount of toxic gas required for a reaction can easily be controlled by adjusting the amount of the surrogate, which is in contrast to the alternative that usually requires a large excess of the toxic gas itself. These advantageous features of toxic gas surrogates contribute to guaranteeing high levels of safety, as well as efficiency when used in organic reactions.
To date, many types of compound are known to be toxic gas surrogates. However, most of them require somewhat harsh conditions and are expensive for use in synthesis. In addition, reactions using these surrogates often suffer from low yields, low reproducibilities, and narrow substrate scopes. Therefore, the development of novel toxic gas surrogates that are cost effective and highly reactive is strongly desired. If such surrogates were at hand, they would not only establish simple and practical reaction protocols adoptable by all chemists, but also accelerate the development of new reactions using toxic gases.
Since we became interested in the advantages of toxic gas surrogates, we have investigated the development of novel ones that are highly reactive, but sufficiently stable, and can be used to realize safe and practical organic reactions. Described herein are our efforts toward the creation of novel CO surrogates and the development of a series of catalytic carbonylative reactions using these CO surrogates. Furthermore, the use of an SO2 surrogate for the selective syntheses of both sulfonamides and sulfinamides is also described. These reactions are significantly safe and highly practical since they require neither external toxic gases nor specialized experimental equipment.
Some of our research has already been described in our previous accounts.22–24) In this review, we highlight important aspects of our past work, while focusing on recent results in detail.
Hydroesterification is an atom-economical, direct method for the introduction of an ester moiety into an alkene or alkyne.3,4,25) This synthetically important transformation is catalyzed by transition metals such as Ni, Ru, and Pd. The pioneering work of Reppe,26) who used acetylene, CO, and an alcohol to afford an ester, has been industrially applied to the synthesis of many kinds of chemical ingredients.3,27) Methyl formate was later found to play an alternative role as a source of CO and methanol.28) However, these synthetic methods are not practical since they require high temperatures and pressure-resistant apparatus.
The reaction conditions were improved in 2002 when the Ru-catalyzed hydroesterification of alkenes using 2-pyridylmethyl formate was reported29–33) (Chart 1A). Two products (1, 2) were obtained, depending on the regioselectivity, which was predominantly affected by the steric properties of the substrate.34) While this reaction could be performed under ambient conditions, 2-pyridylmethyl formate was the only applicable formate. Intramolecular coordination from the nitrogen atom of the pyridine moiety to the Ru center was assumed to play an important role in promoting this reaction. While the Pd-catalyzed hydroesterifications of alkynes using phenyl formate have been reported,35) few examples using alkenes as substrates have been demonstrated, suggesting room for improvement.
We became interested in why such an important reaction had not been generally used to synthesize esters from alkenes. We initially envisaged that ester exchange to form 2-pyridylmethyl formate would facilitate the hydroesterification of a variety of formates; a subsequent second ester exchange would then afford products 3 and 4 (Chart 1B). The exploration of the use of additives during the hydroesterification of 4-methoxystyrene (5) and benzyl formate (6) in the presence of the Ru3(CO)12 catalyst identified effective additives, such as imidazoles 9 and 10. Because 10 cannot undergo ester exchange due to the absence of a hydroxy group, these effective additives might act as ligands to Ru metal, rather than through the initially assumed reaction mechanism36–38) (Chart 1C).

After extensive screening of over 50 ligand candidates, imidazole-type ligands were revealed to be effective, with 13 exhibiting the best performance on the basis of high yields of the desired products36,39) (Table 1). It was noted that only 13 showed a reversal in regioselectivity, although the reasons remain unclear. Bulkier ligands tended to prefer the linear product 7, which suggested that the regioselectivity could be controlled by the steric bulk of the ligand used.
 |
a) Combined yield of 7 and 8. b) Ratio determined by 1H-NMR spectroscopy.
Further optimization of the reaction conditions led to the observation that solvent-free conditions tended to provide higher product yields. The substrate scope (both alkene and formate) was then examined under the optimal conditions, showing that the Ru–imidazole catalyst system greatly improved the scope of the reaction, from the perspectives of both the alkene and formate, compared to the previously reported conditions36,39) (Table 2). Alkyl and aryl formates could be used, with the former affording products in higher yields than the latter (entries 1, 2). Monosubstituted alkenes were good substrates, and the 1,1-disubstituted alkene only afforded the linear product in moderate yield, indicating that steric factors determined both reactivity and regioselectivity (entries 3–5). While cyclic 1,2-disubstituted alkenes such as norbornene and indene reacted well, stilbene as an acyclic example required high temperature conditions due to its low reactivity (entries 6–8).
 |
a) Combined yield. b) Major isomer : minor isomer. The ratio was determined by 1H-NMR spectroscopy. c) Ar=4-MeOC6H4. d) 155°C.
The Ru–imidazole catalyst system also worked well for intramolecular hydroesterifications to give lactones36,40) (Table 3). Interestingly, these reactions preferentially afforded the smaller ring-sized lactone regardless of the structure of the substrate.
 |
a) Combined yield. b) Major isomer : minor isomer. The ratio was determined by 1H-NMR spectroscopy. c) The diastereomeric ratio of the minor isomer was 81 :19.
Furthermore, catalytic intramolecular hydrocarbamoylations41–43) using formamides as substrates afforded lactams in high yields39) (Chart 2). Some oxindoles and related compounds were also obtained regioselectively without difficulty.

To obtain mechanistic insight into hydroesterifications catalyzed by the Ru–imidazole catalyst system, we attempted to isolate a catalyst complex. After much trial and error, we eventually isolated crystals of Ru–18 suitable for X-ray crystallographic analysis (Chart 3A); Ru–18 was revealed to be a 1 : 2 complex of Ru3(CO)12 and 18, bridged such that the nitrogen atom of the imidazole and the oxygen atom of the hydroxymethyl group are connected to different Ru atoms. Hydroesterification reactions using a catalytic amount of isolated Ru–18 exhibited similar reactivity and selectivity to those catalyzed by Ru3(CO)12 and 18 (Chart 3B), which suggests that the Ru–imidazole complex is formed in situ by the reaction of Ru3(CO)12 and an imidazole ligand, and that the complex might be an active catalyst or its precursor.

When the substrate scope of the Ru-catalyzed hydroesterification was examined, we often observed the formation of an alcohol by-product, which possibly came from the Ru-catalyzed decarbonylation of the formate.44) To confirm whether hydroesterification proceeds directly through the formate or via decarbonylation of the formate, the catalytic hydroesterification of 5 was conducted using the 13C-labeled formate 6-13C (Chart 4). While the desired products (7-13C and 8-13C) were obtained in high yield, at most 60% of the 13C label was incorporated into the two products. This result supports the notion that the CO ligand in Ru3(CO)12 can be used as the carbonyl source for hydroesterification, and that the reaction proceeds via decarbonylation of the formate followed by recarbonylation of the Ru intermediate. The catalyst (5 mol% Ru3(CO)12) can release 0.6 equiv of unlabeled CO, and 6-13C is a source of 1.5 equiv of 13C-labeled CO supplied through formate decarbonylation; the observed 13C incorporation ratio was therefore lower than the statistical value (71%). This is ascribable to low levels of 13CO during the initial stage of the reaction.

The existence of the decarbonylation–recarbonylation pathway was further confirmed by performing the hydroesterification with benzyl alcohol instead of benzyl formate (Chart 5). The carbonyl moiety was incorporated into products 7 and 8, indicating that CO insertion into the Ru–OBn bond occurred at some stage during the catalytic cycle.

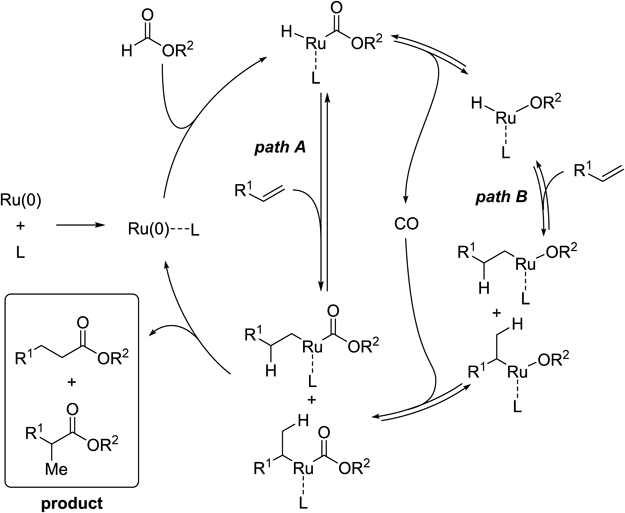
With this mechanistic insight in hand, the proposed reaction mechanism for hydroesterifications catalyzed by the Ru–imidazole catalyst system is shown in Chart 6. Oxidative addition of the C–H bond of the formate affords the acylruthenium intermediate, which then reacts with the alkene either via path A or path B; the latter, involving decarbonylation and recarbonylation processes, has been experimentally confirmed. Two regioisomeric products are finally obtained via reductive elimination that closes the catalytic cycle. At this moment, path A, involving the oxidative addition of the formyl C–H bond to Ru followed by alkene insertion and reductive elimination, cannot be ruled out.
As mentioned in the previous section, the hydroesterification catalyzed by Ru–imidazole is accompanied by the formation of various amounts of alcohol by-products. Among the formates, phenyl formate (20a) produced a significant amount of phenol, resulting in low yield of the hydroesterification product (see Table 2, entry 2). The formation of phenol or other alcohols implies that decarbonylation occurs in the presence of the Ru catalyst, which led us to consider the potential of 20a as a CO surrogate. To realize this potential, we designed a tandem transformation involving formate decarbonylation followed by the Pd-catalyzed alkoxycarbonylation of bromobenzene (19a) without the use of an external source of CO gas (Table 4). This reaction affords the same product as the corresponding Heck alkoxycarbonylation, which was originally performed using CO gas in an alcohol solvent.45)
 |
a) 13 h.
Unless otherwise noted, all reactions mentioned from here onwards were performed in a screw-capped 10-mL test tube (Chart 7). The reaction protocol avoided the use of CO gas from a cylinder; instead it relied on the CO generated by the reaction of the CO surrogate, which is highly safe and practical. A quick investigation of this tandem reaction provided important results; (1) the reaction proceeded even in the absence of a Ru catalyst (entry 2),46) and (2) only phenyl formate afforded the desired phenyl ester, although alkyl formates completely failed (entries 3–5). Furthermore, confirmation of the reaction conditions required for CO generation from 20a (Table 5) revealed a surprising observation; mild heating and (even a catalytic amount of) a weak base such as triethylamine (NEt3) were the only requirements.47) We also noted that NEt3-mediated CO generation was significantly favored over Pd-catalyzed CO generation, which was almost negligible in comparison (entry 2).

 |
a) Yield of phenol determined by HPLC. b) 6.5 h.
At the time of this important discovery of CO generation from 20a, various CO surrogates were known, including formic acid,48–50) paraformaldehyde,50) N,N-dimethylformamide (DMF),51) chloroform,52) aldehydes,53–56) metal carbonyl complexes,57) acid chlorides,58) and silacarboxylic acids.59) Alkyl formates60) had been also reported to act as CO surrogates. However, all of these surrogates have drawbacks that limit their use in synthetic organic reactions due to the harsh conditions required for CO generation and, in some cases, issues of economy and the formation of hazardous waste materials. On the other hand, 20a is easily synthesized and stored, and generates CO under incredibly mild conditions. Since transition metal-catalyzed carbonylation is a fundamental transformation that introduces an additional carbon atom,61–63) the use of 20a as a CO surrogate provides a complementary and convenient method for the preparation of a variety of carbonyl compounds.
3.2. PhenoxycarbonylationFollowing the optimization of the conditions for the phenoxycarbonylation of bromoarenes using 20a as a source of both CO and phenol, the substrate scope of the reaction was examined47,64) (Table 6). We were pleased to find its applicability to a wide range of substrates, including sterically hindered ortho-disubstituted bromoarenes, alkenyl and allyl bromides, and bromoheteroarenes. Except for the substrate bearing the ortho-nitro substituent, the reaction proceeded irrespective of the substrate substituent.
 |
a) β-Bromostyrene (E/Z=87 : 13) was used as a substrate.
Alkenyl tosylates are generally stable, crystalline compounds that are easily prepared from the corresponding carbonyl compound and tosyl chloride. Phenoxycarbonylation was performed using alkenyl tosylates as substrates65,66) (Table 7). While slightly harsh conditions were required, these reactions afforded a variety of α,β-unsaturated esters. Furthermore, one-pot reaction sequences to afford amides were feasible through the addition of the required amine following the phenoxycarbonylation step (Chart 8). These reactions offer convenient synthetic routes to α,β-unsaturated carbonyl compounds.
 |
a) HCO2Ph (4.0 equiv) and Net3 (4.0 equiv) were used.

Another example of the application of phenoxycarbonylation using 20a is the direct conversion of axially chiral ditriflates into the corresponding dicarboxylates67) (Table 8). Some axially chiral carboxylic acids and their derivatives are known to be useful organocatalysts or their precursors.68–70) However, only few synthetic methods have been reported for the construction of binaphthyldicarboxylic acid in short steps.71,72) We optimized the double phenoxycarbonylation of binaphthyl ditriflate, which was easily obtained from commercially available and optically pure 2,2′-binaphthol in a single step. While low yields of the product were obtained, probably due to steric congestion around the two reactive sites, the combination of 1,3-bis(diphenylphosphino)propane (DPPP) and ethyldiisopropylamine (iPr2NEt), that was reported previously by Takaya and colleagues,71) was found to be effective for promoting this double phenoxycarbonylation under neat conditions. This reaction was successfully applied to the phenoxycarbonylation of other axially chiral ditriflates; hence, this procedure constitutes a general method for the synthesis of axially chiral dicarboxylates.
 |
To improve substrate compatibility, the ability to perform these phenoxycarbonylations under milder conditions to those mentioned above is desirable. Accordingly, we turned our attention to enhancing the reactivity of phenyl formate. Considering that alkyl formates failed to react with bromobenzene in the Pd-catalyzed phenoxycarbonylation (see Table 4, entries 4, 5), an aryl group is required for the CO generation from a formate. Actually, CO generation from an alkyl formate in the presence of NEt3 has seldom been observed, even at elevated temperatures. We proposed that the introduction of an electron-withdrawing group on the benzene ring of phenyl formate would accelerate the CO generation. While no detailed mechanistic information regarding CO generation existed at that time, the reaction seemed to proceed via the abstraction of the formyl hydrogen by NEt3 or by nucleophilic attack of NEt3 at the carbonyl carbon. Both pathways would be promoted by decreasing electron density at the formyl group. Furthermore, a reported computational study indicated that the introduction of an electron-withdrawing group on the benzene ring of phenyl formate contributes to a decrease in the bond dissociation energy of the formyl C–H bond.35)
To confirm this hypothesis, several aryl formates bearing electron-withdrawing groups were synthesized, and their abilities to generate CO were examined (Table 9). As expected, faster reactions were observed with increasing electron-withdrawing properties. In particular, 2,4,6-trichlorophenyl formate73) (20h) exhibited high reactivity; its CO generation was complete within 30 min at room temperature; it is also a highly crystalline solid.74) Surprisingly, 20h was found to be extremely stable, as evidenced by 1H-NMR spectroscopy, which confirmed that a sample that had been prepared and stored for over 1 year under ambient conditions was identical to that freshly synthesized.
 |
a) Determined by 1H-NMR.
Fast CO generation from 20h prompted us to examine Pd-catalyzed aryloxycarbonylations of iodoarenes at room temperature (Table 10). To our delight, these reactions proceeded smoothly to afford the corresponding 2,4,6-trichlorophenyl esters in high yields.74) We observed wide substrate scope for this reaction, although slight heating was required for sterically hindered substrates. Alkenyl triflates and bromides, heteroaryl iodides, and even electron-deficient 2-bromopyrimidine were successfully transformed by this reaction.
 |
a) Reaction was performed at 45°C. b) A substrate bearing a triflyloxyl group instead of an iodo group was used. c) A substrate bearing a bromo group instead of an iodo group was used.
The products obtained in these reactions were trichlorophenyl esters bearing three electron-withdrawing chloro groups. These compounds are suitable substrates for nucleophilic attack at the carbonyl group leading to a variety of carbonyl compounds. We attempted several transformations involving these trichlorophenyl esters, and obtained numerous carbonyl compounds, including a carboxylic acid, an alkyl ester, a thioester, and amides (Chart 9). Since the starting trichlorophenyl ester was stable under ambient conditions, the sequence involving aryloxycarbonylation followed by nucleophilic attack is an attractive synthetic strategy for preparing carboxylic acid derivatives from iodoarenes.

The observation that the introduction of an electron-withdrawing group enhances the reactivity of an aryl formate toward NEt3 moved us to investigate the reaction mechanism of aryl formates during the generation of CO and phenol derivatives.75) A further understanding of the mechanism would provide opportunities to develop significantly useful novel CO surrogates with high reactivities, thereby improving carbonylation conditions and enhancing safety and practicality.
Several mechanistic insights into the CO generation from 20a were obtained experimentally. (1) Kinetic studies were performed by reacting 20a with 1.0 equiv of tributylamine (NBu3) at 80°C, leading to the following rate equation:
 |
In parallel with experimental investigations, the TS and intrinsic reaction coordinate (IRC) in the reaction of 20a and trimethylamine (NMe3) was theoretically calculated using density functional theory (DFT); these calculations revealed the existence of two TS structures that depended on the initial conformation of 20a, both of which led to CO and phenol as the final products (Chart 10). In each TS structure, the H–C–O (carbonyl) angle of the formyl group was wider than that of the original structure, indicating that the character of the formyl carbon was changing from sp2 to sp. Notably, the abstraction of the formyl hydrogen in 20a by NMe3 and the elimination of CO and phenoxide occurred simultaneously from each TS.

Based on the combination of experimental and theoretical studies, the proposed mechanism for the CO generation from 20a is presented in Chart 11. The reaction commences with the abstraction of the formyl hydrogen in 20a by the amine acting as a Brønsted base, and this is followed by the simultaneous elimination of CO and phenoxide. The resulting ammonium and phenoxide species then undergo an acid-base reaction to regenerate the amine. This reaction mechanism can be regarded to be a concerted E2 α-elimination, with the amine acting as the catalyst.

The detailed understanding of the reaction mechanism allowed us to control the rate of CO generation by adjusting the reaction conditions. This is an important aspect for successful carbonylation because (1) CO generation and Pd-catalyzed carbonylation are mutually independent processes, and (2) an appropriate rate of CO supply realizes the smooth promotion of the Pd-catalyzed carbonylation step because excessive ligation of CO to Pd, which retards the reaction,76) is suppressed. Our mechanistic findings were used to improve the reaction conditions of Pd-catalyzed phenoxycarbonylations using 20a (Chart 12). Through the use of a polar solvent and a relatively strong base, CO generation from 20a and the phenoxycarbonylation of iodoarenes and electron-deficient bromoheteroarenes that previously required 80°C were realized at room temperature.

Since aryl formates were discovered to be potent CO surrogates, they were then applied to the syntheses of carbonyl compounds other than esters to demonstrate their broad applicability. All of the reactions mentioned above afforded products containing CO and phenol moieties from the starting aryl formates. We envisaged that the introduction of an additional nucleophile would provide a variety of carbonyl compounds via the nucleophilic substitution of aryl esters as aryloxycarbonylation products or via the direct substitution of acylpalladium intermediates (Chart 13).

However, we were concerned that aryl formates would react with nucleophiles to produce other formic acid derivatives rather than generate CO. In fact, secondary amines such as morpholine and dibenzylamine readily reacted with phenyl formate to form amides incapable of generating CO under weakly basic reaction conditions. Subsequently, hydrosilanes were chosen as nucleophiles because these hydrides are mildly nucleophilic and do not undergo undesired reactions with aryl formates. Furthermore, the use of hydrosilanes in Pd-catalyzed carbonylations, namely reductive carbonylations,62,77,78) affords aldehydes from a variety of bromoarenes, and is a powerful synthetic method because it is independent of the electronic nature of the bromoarene.
The reductive carbonylation of 1-bromo-4-methoxybenzene (19b) was examined in the presence of triethylsilane (HSiEt3) using aryl formates as CO surrogates (Table 11). However, all of the aryl formates examined, and acetic formic anhydride (known to generate a CO surrogate in situ),79) failed to afford the desired aldehyde (entries 1–4). In some cases, the formation of aryloxycarbonylated by-products were observed, which clearly indicates that the phenoxide moiety is a stronger nucleophile than HSiEt3. We then investigated highly electron-deficient formamides in the hope that the amido base generated during CO formation had sufficiently low nucleophilicity. To our delight, N-formylsaccharin (20k) proved to be effective in this reaction, affording the desired aldehyde in 80% yield (entry 5).80)
 |
a) Determined by HPLC. b) HCO2K (2.5 equiv) and Ac2O (1.5 equiv) were used in place of Na2CO3.
20k was formerly used as a germicide81) and a formylating agent for amines.82) It is highly crystalline and stable under ambient conditions, and can be synthesized in just a single-step from saccharin, an inexpensive artificial sweetener. While it cannot be dissolved in non-polar solvents, fast CO generation is observed even at room temperature. Hence, 20k can be regarded to be a readily available and practical CO surrogate.
During the optimization of the reductive carbonylation of bromoarenes we discovered that sodium carbonate (Na2CO3) was the effective base compared with organic bases. This is possibly ascribed to the low solubility of Na2CO3 in DMF, which contributes to the slow supply of CO. With the optimized reaction conditions, the substrate scope of this reductive carbonylation was examined using only 1.5 equiv of 20k as a CO surrogate (Table 12). Gratifyingly, various substrates, including electron-deficient bromoarenes and bromoheteroarenes, afforded the desired products in moderate to high yields. Notably, this reaction was applied to the synthesis of an isotopically labeled compound; 4-phenylbenzaldehyde-d, bearing a deuterated formyl group, was synthesized using triethylsilane-d.
 |
a) Pd(OAc)2 (1.5 mol%), DPPB (2.3 mol%), 20k (1.5 equiv), Et3SiH (1.5 equiv), and Na2CO3 (2.5 equiv). b) Pd(OAc)2 (5 mol%). c) Et3SiD (1.3 equiv) was used instead of Et3SiH.
Monitoring of the reaction mixture by electrospray-ionization-time-of-flight (ESI-TOF)-MS revealed the formation of acylsaccharin, suggesting that the nucleophilic attack by saccharinate generated at the time of CO generation occurred (Chart 14A). In addition, a control experiment revealed that an acylsaccharin is converted into the aldehyde under the reaction conditions of the reductive carbonylation (Chart 14B). With these results in mind, two mechanistic pathways are proposed to operate during reductive carbonylation (Chart 15). The first mechanism involves the formation of the acylpalladium bromide followed by direct anion exchange of the bromide for hydride and reductive elimination (path A). The second mechanism involves the attack of in-situ-generated sodium saccharinate, which is assumed to be sufficiently nucleophilic, at the acylpalladium bromide to form the acylpalladium saccharinate (path B). HSiEt3 then reacts with the acylpalladium saccharinate followed by reductive elimination to afford the product. The acylpalladium saccharinate appears to be in equilibrium with the acylsaccharin intermediate.


While acyl fluorides are attractive synthetic intermediates of carbonyl compounds because they are known to be more stable than their acyl chloride counterparts83) and react with various nucleophiles, no general, catalytic method for the synthesis of acyl fluorides has been developed.84,85) For the further utilization of 20k as a CO surrogate, we developed a Pd-catalyzed fluorocarbonylation of bromoarenes using a fluoride source, with the expectation that the fluoride would act as both the fluorinating agent and a Brønsted base. We did not exclude the fact that the fluoride might react with 20k to form formyl fluoride; however if this was the case, formyl fluoride appeared to react with more fluoride to generate CO because the high electron-withdrawing nature of the fluorine atom increases the acidity of the formyl hydrogen. Although it was not clear which situation operated, the Pd-catalyzed carbonylation followed by fluoride addition was found to be feasible, affording acyl fluorides in moderate yields86) (Chart 16). Since partial hydrolysis was observed during the purification of these acyl fluorides by silica gel column chromatography, additional nucleophiles were introduced after the fluorocarbonylation step, in a one-pot manner, to readily afford carboxylic acid derivatives in high yields (Table 13).

 |
a) NuH=BnNH2 or C12H25SH.
As applications of external-CO-free carbonylation, the carbonylative cyclizations of haloarenes bearing nucleophilic moieties were next examined87) (Chart 17), which gave rise to a variety of cyclic carbonyl compounds. Although some carbonylative cyclization methods have been reported,56,88–90) they generally require pressurized CO gas and high temperature, which hampers the implementation of these reactions. Therefore, the development of a simple and practical method for the synthesis of cyclic compounds is highly desired.

A model reaction using diester 21 as the substrate revealed that polar solvents such as DMF and dimethyl sulfoxide (DMSO) are effective, which is ascribable to acceleration of the nucleophilic cyclization step (Table 14). The reaction can also be conducted at a relatively low temperature (80°C) compared to previously reported methods. We were pleased to find that carbon-, nitrogen-, and oxygen-based nucleophiles were applicable to this reaction to afford various classes of cyclic carbonyl compounds that demonstrated the wide substrate scope of this reaction (Table 15).
 |
a) Isolated yield of 22. Isolated yield of by-product 23 is shown in parehthesis. b) NMR yield.
 |
a) P(t-Bu)3·HBF4 (15 mol%) was used instead of Xantphos. b) NMR yield. c) DMF was used as the solvent.
Carbonylative reactions using CO surrogates are advantageous from the viewpoints of safety and practicality. These features are favorable for conducting carbonylations on both laboratory and industrial scales. It is important to show that large-scale carbonylations are feasible in order for the method to become widely accepted in the chemistry community.
Typical reactions were conducted on a 0.25 mmol scale during the above-mentioned investigations into these carbonylative reactions, and a 10-mL screw-capped test tube with a silicone rubber septum was used as the reaction vessel. The internal pressure of the test tube appeared to increase as the CO surrogates reacted to generate CO. In large-scale syntheses, extra care needs to be paid to this internal pressure because the generation of a large volume of CO inside the vessel sharply increases the risk of an explosion. This pressure issue was solved through the attachment of an empty balloon directly to the vessel that ensures that the internal pressure is maintained at 1 atm.
The gram-scale phenoxycarbonylation of binaphthyl ditriflate (24) using 20a was then conducted67) (Chart 18A). While inflation of the balloon was observed during the reaction, the internal pressure was continuously maintained at 1 atm. The reaction proceeded to afford 1.24 g of the desired product 25 (63% yield). Another example of a gram-scale reaction using 20a is the carbonylative cyclization of 26 (Chart 18B) that successfully afforded 1.77 g of 27 (70% yield), which is a bioactive compound.87)
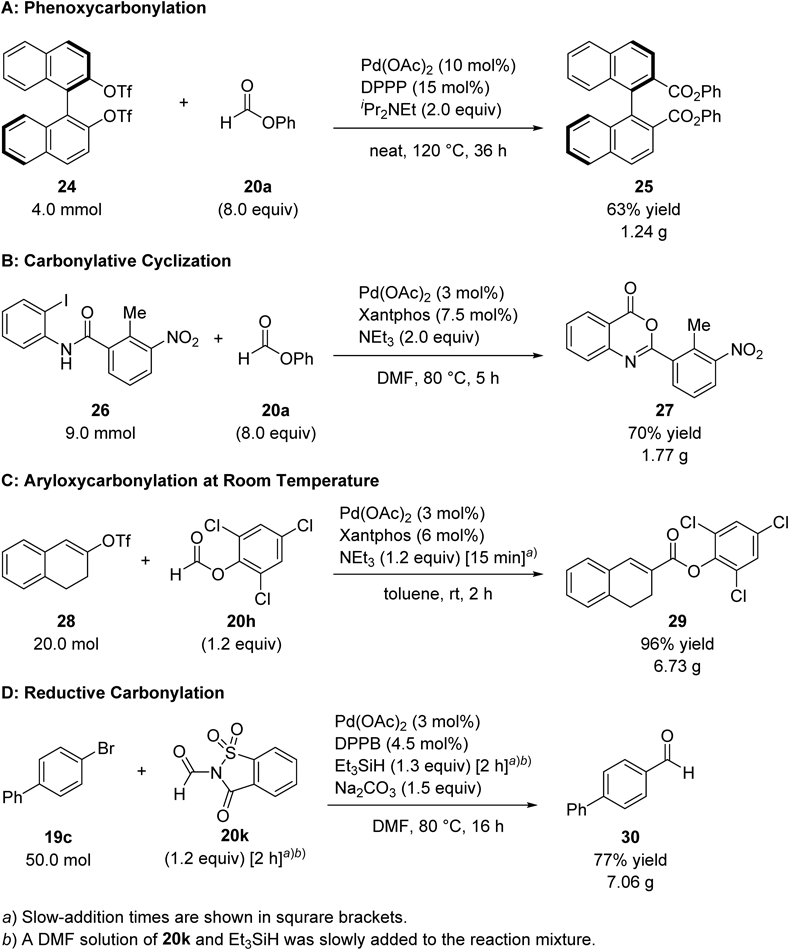
As for the use of other CO surrogates in gram-scale carbonylative reactions, 20h was applied to the aryloxycarbonylation of alkenyl triflate 28 on a 20 mmol scale91) (Chart 18C); more than 6 g of the desired 2,4,6-trichlorophenyl ester 29 was obtained in a single run. In this case, the slow addition of NEt3 using a dropping funnel was necessary to avoid the rapid generation of excess CO that might retard the reaction by suppressing the oxidative addition of 28 (Chart 19). This slow addition strategy was also effective for the gram-scale synthesis of aldehyde 30 from bromoarene 19c using 20k92) (Chart 18D). A solution of 20k and HSiEt3 was slowly added to the reaction mixture in order to prevent the overly rapid generation of CO and the formation of the reduced by-product of 19c by HSiEt3. Applying the slow addition technique, 7 g of the desired aldehyde 30 was obtained in a single run on a 50 mmol scale.

Sulfonamides are an important class of compound because they are found in many drugs.93,94) The syntheses of sulfonamides have mostly relied on the condensations of the required sulfonyl chlorides and amines.95) However, sulfonyl chlorides are difficult to handle due to their corrosivenesses and susceptibilities toward hydrolysis. With limited commercial access to sulfonyl chlorides, multi-step preparations from sulfonic acid are required in some cases.96)
Since we are interested in the use of toxic gas surrogates in synthetic organic chemistry, we envisaged the formation of sulfonamides via the direct three-component reactions of haloarenes, an SO2 surrogate, and amines. We also expected that the mode of SO2 coordination to the transition metal catalyst would be similar to that of CO, which further motivated our investigation into sulfonamide chemistry.97) Some papers reported a similar concept for the synthesis of sulfonyl hydrazides in which hydrazines were used instead of amines98,99) (Chart 20A); however, these synthetic methods never afforded sulfonamides. Since transition metal-catalyzed insertion of a sulfonyl group using SO2 gas97) or its surrogate15–17) is a well-known process, the failure to synthesize sulfonamides is presumably due difficulties associated with the nucleophilic attack of the amine at the in-situ-generated sulfonyl moiety. Therefore, we decided to use haloarenes bearing amine moieties with the expectation that cyclic sulfonamides would be obtained via the transition metal-catalyzed insertion of SO2 derived from its surrogate, followed by entropically favored cyclization to promote the difficult N–S bond forming step100) (Chart 20B). This synthetic strategy was motivated by the successful development of the catalytic carbonylative cyclization using phenyl formate.87)

An attempt to convert model substrate 31 into a cyclic sulfonamide using potassium metabisulfite (K2S2O5), an inexpensive SO2 surrogate,99) under palladium catalysis is shown in Chart 21. An initial study revealed that DMSO was the only solvent that afforded the desired cyclic sulfonamide 32. To our great surprise, when we added tributylamine (NBu3) to capture the in-situ-formed hydrogen iodide, only sulfinamide 33, the mono-deoxygenated analogue of 32, was obtained with complete selectivity. Sulfinamides are used as chiral auxiliaries101) and chiral ligands102) in synthetic chemistry, as well as amide isosteres in peptidomimetics.103) These initial results inspired us to focus on the development of selective syntheses of both sulfonamides and sulfinamides.

Optimization of the reaction conditions revealed that the most influential factors that determined product selectivity were the base and the amount of base used (Table 16). With 1.0 equiv of base, a mixture of sulfonamide 32 and sulfinamide 33 was obtained in most cases (entries 1–3). Among the various bases, only NBu3, as a less-hindered tertiary amine, afforded sulfonamide 32 selectively (entry 5). As the amount of NBu3 was varied, a striking product-selectivity effect was observed; 1.0 equiv afforded the highest yield of sulfonamide 32, and 2.0 equiv gave the highest yield of sulfinamide 33 (entries 4–8).
 |
Monitoring of the reaction revealed that sulfinamide 32 is the common initial product in reactions that afforded both sulfonamide 32 and sulfinamide 33 (Chart 22). During the synthesis of 32, in-situ-formed 33 was gradually converted into 32 immediately upon complete consumption of substrate 31. On the contrary, such a conversion was never observed when the reaction conditions for the selective synthesis of 33 were applied. These results strongly indicated that K2S2O5 is an “S=O” source that introduces the sulfinyl group, and that oxidation of the sulfinamide to the sulfonamide depends on the reaction conditions.

After optimizing the reaction conditions, the substrate scope for these selective syntheses of sulfonamides and sulfinamides was examined (Table 17). Both sulfonamides and sulfinamides were obtained in moderate to good yields regardless of the positions and electronic properties of substituents on the benzene ring. While the N-phenyl substrate failed to selectively afford the sulfinamide, other substrates exhibited almost complete selectivity. The substrate bearing the pyridine ring and that with a longer carbon tether were difficult to convert into the corresponding desired product. We are currently improving the substrate scope of this reaction; investigations into the reaction mechanism are also currently underway.
 |
a) Yield of sulfonamide. b) 120°C. c) Complex mixture.
Several novel carbon monoxide (CO) surrogates and efficient organic reactions using toxic gas surrogates were successfully developed, allowing these reactions to be conducted in a highly safe and practical manner. When we focused on the use of formates as C1 sources, hydroesterification of alkenes using formates were found to be catalyzed by the Ru–imidazole catalyst system. This catalyst greatly expanded the substrate scope, and mechanistic studies revealed the presence of a reaction pathway that involved the decarbonylation of the formate and the recarbonylation of the Ru intermediate by a carbonyl ligand. The observation of alcohol by-products promoted by Ru catalysis, which was especially prominent in the case of phenyl formate, led us to discover that phenyl formate generates CO by reaction with a weak base under mild conditions. This discovery provided an important opportunity to create formic acid derivatives as reactive but stable CO surrogates, and to develop a series of room-temperature aryloxycarbonylation reactions. Experimental and theoretical mechanistic studies into the CO generation from phenyl formate indicated that CO is generated through a concerted E2 α-elimination process; the elucidation of this reaction mechanism led to the establishment of more efficient and milder reaction conditions for catalytic phenoxycarbonylations. Furthermore, catalytic carbonylation using N-formylsaccharin as a stable but highly reactive CO surrogate facilitated the use of nucleophiles, providing access to a variety of carbonyl compounds. We demonstrated several carbonylative reactions on the gram scale using our CO surrogates, highlighting their further potential in terms of scalability and practicality. As for the use of other toxic gas surrogates, the selective syntheses of sulfonamides and sulfinamides, using potassium metabisulfite (K2S2O5) as a sulfur dioxide surrogate, was achieved for the first time. The choice of base and the amount of base used determined selectivity, with K2S2O5 regarded to be an “S=O” source.
Organic reactions using toxic gas surrogates simply require the reagents to be mixed. Importantly, all reactions mentioned in this review using toxic gas surrogates were conducted without the use of an external source of toxic gas or special pressure-resistant apparatus, rendering their experimental protocols significantly simpler and more practical than those of conventional reactions using pressurized gases. In addition, since some CO surrogates developed in our laboratory are now commercially available at reasonable prices,104) they have begun to be applied in the fields of medicinal chemistry,105,106) total syntheses of natural bioactive compounds,107,108) and flow chemistry.109,110) For the development of truly useful reactions, the practical aspects of the organic reactions in question need to be considered during their development and application. The substitution of toxic gases for their surrogates in synthetic organic reactions is attractive; the resulting ready-to-use reactions may result in paradigm-shifting protocols for conducting reactions with toxic gases. Furthermore, the development of novel toxic gas surrogates and their use in synthetic organic reactions will potentially open new fields of chemistry involving toxic gases, while enabling rapid and highly versatile derivatization processes. These developments will contribute to a dramatic shortening in the time required for the research and development of functional materials and drug discovery.
This review of the author’s work was written by the author upon receiving the 2017 Pharmaceutical Society of Japan Award for Young Scientists.
All of the research in this review was conducted at the School of Pharmaceutical Sciences, University of Shizuoka. I would like to express my sincere appreciation to Professor Kei Manabe (University of Shizuoka) for his continuous support and fruitful discussions. I am grateful to Professor Hiroaki Tokiwa (Rikkyo University), Associate Professor Yoshinobu Ishikawa (University of Shizuoka), Professor Hiroshi Hashimoto (University of Shizuoka), and Professor Yasuteru Shigeta (Tsukuba University) for kind advice and helpful support in relation to the theoretical calculations. Dr. Tsuyoshi Ueda (Daiichi-Sankyo Co., Ltd.) is acknowledged for his critical contribution to the initial studies involving CO surrogates. I am also appreciative to all the student co-workers whose names appear in our publications cited in this review. This work was supported by a Grant-in-Aid for Young Scientists (B) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan; Platform for Drug Discovery, Informatics, and Structural Life Science from MEXT; Japan Agency for Medical Research and Development (AMED); Uehara Memorial Foundation; Takeda Science Foundation; and Daiichi-Sankyo, Co., Ltd.
The author declares no conflict of interest.