Fluorine-containing organic molecules play an important role in medicinal chemistry, biological studies, and material sciences due to the unique properties of fluorine: high electronegativity and small size. In the field of medicinal chemistry and drug discovery, the introduction of fluorine atoms into a molecule may improve metabolic stability, solubility, and biological activity. Up to 20% of pharmaceuticals are estimated to contain fluorine.1–3) The development of effective method for synthesizing fluorine-containing functional molecules has thus gained widespread significant attention in organic synthesis.4–11)
Direct functionalization of inert C–H bonds12–15) is a powerful tool to realize atom-16) and step-economical17) synthesis, and fluorine or fluorine-containing small functional groups are successfully introduced with various catalysts and reagents.4–9) More recently, C–H functionalization reactions accompanied by selective C–F bond cleavage of multi-fluorinated coupling partners were explored. Loh and colleagues reported monofluoroalkene synthesis using gem-difluoroalkenes via aromatic C–H activation under Cp*Rh(III) (Cp*=pentamethylcyclopentadienyl) catalysis to afford monofluoroalkenes,18) which can serve as peptide mimics19,20) (Chart 1a). They proposed that β-F elimination after C–H bond cleavage and the insertion of C–C double bonds furnishes the product. Once they demonstrated the viability of this C–H/C–F bond cleavage strategy, the scope was rapidly expanded.21–26) A Cp*Co(III) catalyst,27–31) which is more available than Cp*Rh(III)32–34) but often exhibits similar catalytic activity, was used by Li and colleagues21) and Ackermann and colleagues22) for the same transformation. Manganese catalysts were also effective, as reported by Ackermann’s group23) and Feng and Loh’s group.24) Not only the alkenylation, but also allylation using allyl fluorides were also reported by Ackermann22,23) (Chart 1b). Although these reactions are very attractive due to the availability of the fluorinated reagents, the scope of the substrate involved in the C–H activation is still limited to highly reactive indoles and rather simple aromatic compounds.
6-Arylpurines have anti-mycobacterial, anti-hepatitis C virus (HCV), and cytostatic activities,35–38) and the purine moiety works as a good directing group for C–H bond activation with transition metal catalysts.39–49) During the course of our investigation of Cp*Co(III)-catalysis,47,48,50–54) we became interested in catalytic C–H bond functionalization of 6-arylpurine derivatives, and recently reported C–H allylation47) and trifluoromethylthiolation48) reactions. In this communication, we report C–H bond monofluoroalkenylation using gem-difluoroalkenes and allylation using allyl fluorides under Cp*Co(III) catalysis (Chart 1, this work).

Chart 1. Synthesis of Monofluoroalkenes via C–H/C–F Bond Cleavage
We first optimized the reaction conditions for N-isopropyl-6-phenylpurine 1a and gem-difluoroalkene 2a (Table 1). Solvent screening using 5 mol% of [Cp*Co(CH3CN)3](SbF6)2 revealed that 2,2,2-trifluoroethanol (TFE) and 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP), highly-polar but weakly-coordinating fluorinated alcohols, were suitable for this reaction (entries 4, 5), giving the desired fluoroalkene 3aa in excellent yields, while less polar solvents led to lower reactivity (entries 1–3). A dicationic cobalt catalyst generated in situ by using Cp*Co(CO)I2 and AgSbF6 afforded the slightly lower yield (entry 6). We selected the single component catalyst [Cp*Co(CH3CN)3](SbF6)2 for further investigation due to the simpler experimental operation and the better reactivity. Lowering the reaction temperature decreased the yield (entries 7, 8), and 80°C was determined to be optimal. Finally, product 3aa was isolated in 85% yield after silica gel column chromatography (entry 9). The reaction proceeded with high (Z)-selectivity, and 3aa was obtained as an almost single isomer.
Table 1. Optimization of Reaction Conditions Using 6-Arylpurine
1aa) |
|---|
| Entry | Catalyst (X) | Solvent | T (°C) | % Yieldb) |
|---|
| 1 | [Cp*Co(CH3CN)3](SbF6)2 (5) | DCE | 80 | 59 |
| 2 | [Cp*Co(CH3CN)3](SbF6)2 (5) | Toluene | 80 | 10 |
| 3 | [Cp*Co(CH3CN)3](SbF6)2 (5) | THF | 80 | 51 |
| 4 | [Cp*Co(CH3CN)3](SbF6)2 (5) | TFE | 80 | 77 |
| 5 | [Cp*Co(CH3CN)3](SbF6)2 (5) | HFIP | 80 | 95 |
| 6 | Cp*Co(CO)I2 (5)+AgSbF6 (10) | HFIP | 80 | 81 |
| 7 | [Cp*Co(CH3CN)3](SbF6)2 (5) | HFIP | 50 | 36 |
| 8 | [Cp*Co(CH3CN)3](SbF6)2 (5) | HFIP | rt | 9 |
| 9c) | [Cp*Co(CH3CN)3](SbF6)2 (5) | HFIP | 80 | 85d) |
a) The reactions were run using 1a (0.050 mmol), 2a (0.075 mmol), catalyst, and solvent (0.5 mL) under Ar unless otherwise noted. DCE=1,2-dichloroethane. b) Determined by 1H-NMR analysis of the crude mixture using dimethyl sulfone as an internal standard. c) 0.20 mmol scale. d) Isolated yield after silica gel column chromatography.
The scope of the monofluoroalkenylation of 6-arylpurines is summarized in Chart 2. We first evaluated the scope of 6-arylpurines 1. N-Benzyl-protected 6-arylpurines as well as N-isopropyl-protected 6-arylpurines were suitable substrates under the optimized conditions. Both electron-donating groups and electron-withdrawing groups are compatible on the aryl moiety of purines, but electron-deficient substrates exhibited attenuated reactivity (3da, 3ga, 3 ha). When the aryl group had a substituent at the meta-position, the sterically less hindered C–H bond was selectively functionalized (3 ha). We also investigated the scope of gem-difluoroalkenes 2. Methoxy-, methoxycarbonyl-, and bromo-substituted alkenes provided the products in 49–87% yields (3ab–ae). In all cases, no significant side reaction was observed, demonstrating the good functional group compatibility of the reaction conditions.
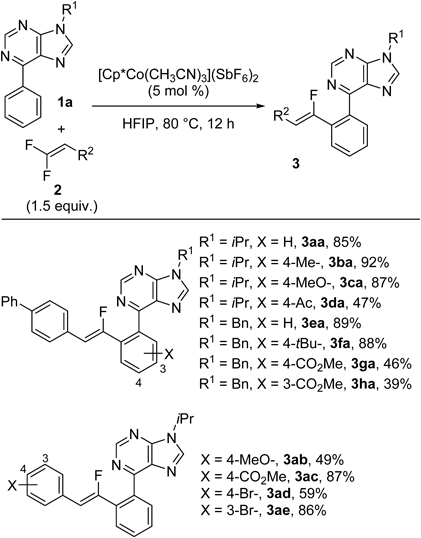
Chart 2. Synthesis of Monofluoroalkenes: Substrate Scope
We next investigated an allylation reaction using allyl fluorides 4 (Chart 3). The reactions proceeded with several substrates of different perfluoroalkyl groups, and products 5aa–ac were obtained in good yields and with moderate Z selectivity.55)

Chart 3. C–H Allylation Using Allyl Fluorides
Finally, we checked the scalability and robustness of the optimized reaction conditions (Chart 4). The reaction of 6-arylpurine 1a and allyl fluoride 4c was carried out in 1.0 mmol scale under air in refluxed TFE, and the product 5ac was isolated in 67% yield. Although the yield was somewhat diminished, the simple setup is more practical for application in medicinal chemistry.

Chart 4. Preparative-Scale Reaction under Air
A plausible catalytic cycle based on previous studies21,22,42–49) is shown in Chart 5. Coordination of 6-arylpurine 1 to a Co(III) species I and subsequent cyclometalation would form intermediate II. In the case of monofluoroalkenylation, insertion of gem-difluoroalkenes 2 to give IIIa and β-fluoride elimination afforded products 3. Allylation would also proceed similarly through intermediate IIIb, which would undergo β-fluoride elimination leading to 5.

Chart 5. Plausible Catalytic Cycle
In summary, we demonstrated that 6-arylpurines underwent Cp*Co(III)-catalyzed C–H monofluoroalkenylation using gem-difluoroalkenes and allylation using allyl fluorides. Both reactions proceeded in moderate to good yields in the presence of a user-friendly single-component catalyst [Cp*Co(CH3CN)3](SbF6)2 in fluorinated alcohol solvents without any additives. Robustness was also demonstrated by a preparative-scale reaction under air. These transformations can increase easily accessible 6-arylpurine derivatives, potentially leading to the discovery of novel biologically active compounds.