Experimental
ChemistryReagents and solvents were obtained from Sigma-Aldrich (St. Louis, MO, U.S.A.). To evaluate the purity of the compounds, uncorrected melting point determination was carried out using Büchi model B-540 melting point apparatus (BÜCHI Labortechnik AG, Flawil, Switzerland). The progress of the chemical reactions as well as the purity of the final products were monitored by TLC using UV and iodine oxidation as revealing agents. The IR attenuated reflectance (ATR) spectra were obtained using a spectrophotometer Perkin ATX-Elmer (PerkinElmer, Inc., Waltham, MA, U.S.A.). 1H-NMR spectra of the synthetized compounds were obtained in dimethyl sulfoxide (DMSO)-d6 solutions at 400 MHz on a Varian MR-400 (9.2 T) instrument. Chemical shift values (δ) were reported in parts per million (ppm) relative to the solvent signal. Mass spectra were obtained using a Thermo-electron model DFS instrument.
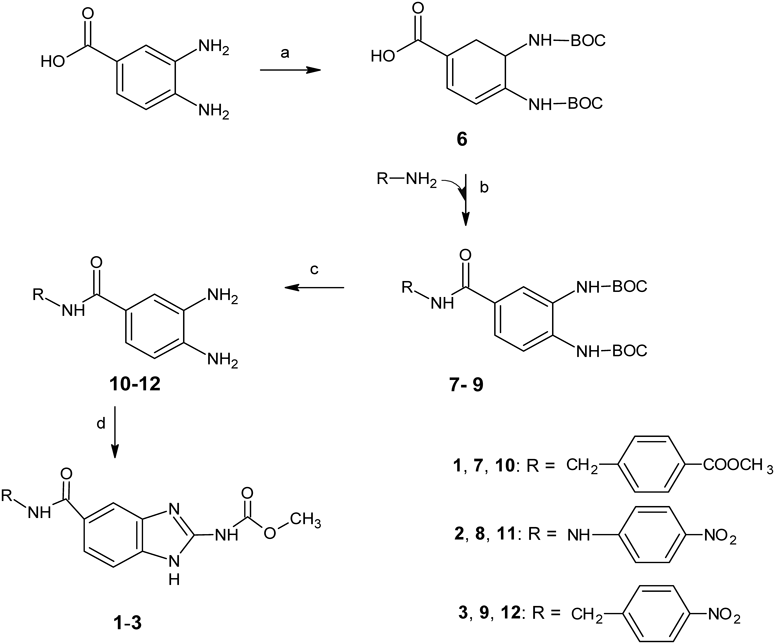
General Procedure for Synthesis of the Benzimidazole Derivatives (1–3)Compounds 7–9 (6.3 mmol, Chart 1) and trifluoroacetic acid (64 mmol) were stirred at room temperature for 5 h. The reactive mixture was then neutralized with saturated NaHCO3 producing a yellow precipitate (10–12). This crude product was filtered and used immediately without purification in the cyclocondensation reaction. A mixture of benzene-1,2-diamine compound (10–12) (6.3 mmol), methanol (25 mL), water (50 mL) and acetic acid (0.5 mL) as a catalyst was heated under reflux for 8 h. Subsequently, 50 mL of ice was added and the mixture was vacuum-filtered to obtain a brown solid, which was then washed with toluene.
Methyl 4-[({2-[(Methoxycarbonyl)amino]-1H-benzimidazol-5-carbonyl}amino)methyl] Benzoate (1)White solid. Yield: 50%. mp: 299°C with decomposition. 1H-NMR (300 MHz, DMSO-d6) δ: 11.98–11.96 (br s, 1H, CONH), 10.47–10.44 (br s, 1H, CONH), 8.27 (d, 2H, H-3′, J = 8.6 Hz), 8.11 (d, 1H, H-4, J = 1 Hz), 7.80 (d, 1H, H-7, J = 8.5 Hz), 7.75 (d, 2H, H-2′, J = 8.6 Hz), 7.49 (dd, 1H, H-6 J6–7 = 8.5, J6–4 = 1 Hz), 4.9 (s, 3H, C-OCH3), 3.80 (s, 3H, N-CO-OCH3). IR (KBr) cm−1: 3331 (NH), 1800 (CONH), 1745 (ArCOOCH3), 1730 (C=O), 1651 (C=O), 1095.08 (C–O–C). MS m/z: 383 (M+). Anal. Calcd for C19H18N4O5 (382.37): C, 59.68; H, 4.74; N, 14.65. Found: C, 59.66; H, 4.72; N, 14.61.
Methyl {5-[2-(4-Nitrophenyl)hydrazinecarbonyl]-1H-benzimidazol-2-yl}carbamate (2)White solid. Yield: 63.9%. mp >300°C. 1H-NMR (300 MHz, DMSO-d6) δ: 10.82 (br s, 1H, CONH), 9.27 (s, 1H, Ar-NH), 8.01 (d, 2H, H-3′, J = 8.6 Hz), 7.97 (d, 1H, H-4, J = 1 Hz), 7.66 (d, 1H, H-7, J = 8.5 Hz), 7.66–7.65 (dd, 1H, H-6, J6–7 = 8.5, J6–4 = 1.0 Hz), 7.72–7.40 (d, 1H, H-7, J = 8.5 Hz), 6.86 (d, 2H, H-2′, J = 8.6 Hz), 3.88 (s, 3H, N-CO-OCH3). IR (KBr) cm−1: 3343 (NH), 1730 (C=O), 1651 (C=O), 1524 (Ar-NO2), 1365 (Ar-NO2), 1095.08 (C–O–C). MS m/z: 371 (M+). Anal. Calcd for C16H14N6O5 (370.31): C, 51.89; H, 3.81; N, 22.69. Found: C, 50.42; H, 3.84; N, 22.79.
Methyl (5-{[(4-Nitrophenyl)methyl]carbamoyl}-1H-benzimidazol-2-yl)carbamate (3)Light-brown solid. Yield: 65.4%. mp: 278.5–278.9°C. 1H-NMR (300 MHz, DMSO-d6) δ: 9.06 (t, 1H, J = 6 Hz, CONH), 8.19 (d, 2H, H-3′, J = 8.6 Hz), 7.97 (d, 1H, H-4, J = 1 Hz), 7.67 (d, 1H, H-7, J = 8.5 Hz), 7.55–7.65 (d, 2H, H-2′, J = 8.6 Hz), 7.41–7.43 (dd, 1H, H-6, J6–7 = 8.5, J6–4 = 1.0 Hz), 4.57 (d, 2H, Ar-CH2-), 3.74 (s, 3H, CO-OCH3). IR (KBr) cm−1: 3373 and 3310 (NH), 1721 (C=O), 1627 (C=O), 1538 (NH), 1519 and 1343.48 (Ar-NO2), 1274 and 1184 (C–O–C). MS m/z: 370 (M+). Anal. Calcd for C17H15N5O5 (369.33): C, 55.28; H, 4.09; N, 18.96. Found: C, 54.47; H, 4.08; N, 18.66.
Synthesis of 3,4-Bis[(ter-butoxycarbonyl)amino]benzoic Acid (6)A mixture of 3,4-diaminobenzoic acid (4 g, 26.2 mmol), 80 mL of water, potassium hydroxide (6.7 g, 0.1051 mmol, 88%), di-tert-butyl dicarbonate (0.1051 mmol) and 60 mL of tert-butanol was stirred at room temperature for 48 h. The reaction mixture was then washed three times with 50 mL of hexane. The aqueous phase was collected in a clean beaker and the mixture was adjusted to pH 4 by the addition of citric acid. After filtration, a light brown solid was obtained (7.8 g) with the following characteristics: Yield: 85%. mp: 199.1–199.7°C. 1H-NMR (300 MHz, DMSO-d6) δ: 12.8 (s, 1H, OH), 8.73 (s, 1H, NH, interchangeable with D2O), 8.68 (s, 1H, NH, interchangeable with D2O), 7.70 (s, 1H, H-2), 7.68 (s, 1H, H-5), 7.62 (dd, 1H, J = 2 Hz, J = 8.4 Hz, H-6), 1.5 (s, 18H, CH3). IR (KBr) cm−1: 3262 and 3310 (–OH and NH), 2975 (–CH3), 1698 (C=O), 1682 (C=O), 1540 (C=C, Ar) and 1157 (C–O–C). MS m/z: 353 (M+). Anal. Calcd for C17H24N2O6 (352.38): C, 57.94; H, 6.86; N, 7.95. Found: C, 57.91; H, 6.85; N, 7.93.
General Procedure for Synthesis of 7–9A mixture of 1,1′-carbonyldiimidazole (CDI, 1.3169 g, 8.1 mmol), compound 6 (2.8618 g, 8.1 mmol) and 35 mL acetonitrile was stirred for 8 h at 60°C. The appropriate benzilamine (methyl 4-(aminomethyl)benzoate, 1-(4-nitrophenyl)methanamine) or 4-(nitrophenyl)hydrazine (8.9 mmol) and N,N-diisopropylethylamine (DIPEA) (1.4 mL, 8.9 mol) were added and the reaction mixture was stirred for 24 h at 80°C. After removing acetonitrile, a reddish-brown oil was obtained. After the addition of 50 mL of water, a light brown solid was obtained in all cases. This solid product was filtered, washed with ether and dried.
Synthesis of Methyl 4-[(3,4-Bis{ter-butoxycarbonylamino} Benzamido) Methyl] Benzoate (7)Yield: 87%. mp: 113–115°C. 1H-NMR (300 MHz, DMSO-d6) δ: 9.18 (t, 1H, J = 6 Hz, CONH), 8.70 (br s, 1H, CONH), 8.19 (d, 2H, H-3′, J = 8.6 Hz), 8.02 (d, 1H, H-5, J = 8.5 Hz), 7.65 (dd, 1H, H-6, J6–5 = 8.5, J6–2 = 1.0 Hz), 7.99 (d, 1H, H-2, J = 1 Hz), 7.60 (d, 2H, H-2′, J = 8.6 Hz), 4.55 (d, 2H, J = 6 Hz, Ar-CH2), 3.75 (s, 3H, CO-OCH3), 1.48 (s, 9H, CH3). IR (KBr) cm−1: 3331 (NH), 1800 (CONH), 1745 (ArCOOCH3), 1730 (C=O), 1651 (C=O), 1095.08 (C–O–C). MS m/z: 500.5 (M+). Anal. Calcd for C26H33N3O7 (499.55): C, 62.51; H, 6.66; N, 8.41. Found: C, 62.52; H, 6.64; N, 8.40.
Synthesis of Di-tert-butyl 5-[(4-Nitrophenyl)hydrazinecarbonyl]-1,2-phenylenebiscarbamate (8)Yield 75%. mp: 130–132°C. 1H-NMR (300 MHz, DMSO-d6) δ: 10.92 (bs, 1H, CONH), 9.20 (s, 1H, Ar-NH), 8.10 (d, 2H, H-3′, J = 8.6 Hz), 8.1 (d,1H, H-4, J = 1 Hz), 7.80 (d, 1H, H-5, J = 8.5 Hz), 7.67 (dd, 1H, H-6, J6–5 = 8.5, J6–2 = 1.0 Hz), 6.88 (d, 2H, H-2′, J = 8.6 Hz), 1.52 (s, 9H, CH3). IR (KBr) cm1: 3287 (–NH), 2979 (–CH3), 1700 (C=O), 1519 (Ar-NO2), 1341 (Ar-NO2), 1241 and 1153 (C–O–C). MS m/z: 488.5 (M+). Anal. Calcd for C23H29N5O7 (487.51): C, 56.67; H, 6.00; N, 14.37. Found: C, 56.69; H, 5.99; N, 14.36.
Synthesis of Di-tert-butyl 5-{[(4-Nitrophenyl)methyl]carbamoyl}-1,2-phenylenebiscarbamate (9)Yield 83%. mp: 110.1–110.6°C. 1H-NMR (300 MHz, DMSO-d6) δ: 9.14 (t, 1H, J = 6 Hz, CONH), 8.69 (s, 1H, CONH), 8.20 (d, 2H, H-3′, J = 8.6 Hz), 8.02 (d, 1H, H-5, J = 8.5 Hz), 7.67 (dd, 1H, H-6, J6–5 = 8.5, J6–2 = 1.0 Hz), 7.99 (d,1H, H-2, J = 1 Hz), 7.58 (d, 2H, H-2′, J = 8.6 Hz), 4.58 (d, 2H, J = 6 Hz, Ar-CH2), 1.48 (s, 9H, CH3). IR (KBr) cm1: 3287 (–NH), 2979 (–CH3), 1700 (C=O), 1517 (Ar-NO2), 1344 (Ar-NO2), 1241 and 1153 (C–O–C). MS m/z: 487.5 (M+). Anal. Calcd for C24H30N40 (486.51): C, 59.25; H, 6.22; N, 11.52. Found: C, 59.36; H, 6.32; N, 11.59.
General Procedure for Synthesis of 10–12Compounds 7–9 (6.3 mmol) and trifluoroacetic acid (64 mmol) were stirred for 5 h at room temperature. The reaction mixture was then neutralized with saturated NaHCO3 producing a yellow precipitate (10–12). After filtration, 10–12 were used immediately without purification in the cyclocondensation reaction to obtain 1–3.
Ethics StatementIn vivo studies were performed according to Mexican Guidance (NOM-062-ZOO-1999). The animals were housed in polycarbonate cages in an environmentally controlled room at 18–23°C and 40–70% relative humidity under a 12 : 12 h light : dark cycle. Food and water were freely available before the start of the experiment.
The protocol (104/12) was approved by the Animal Care Committee of the Instituto Nacional de Neurología y Neurocirugía (Mexico City, Mexico).
In Vitro Cysticidal ActivityStock solutions of each compound (2 mg/mL) and the positive control, albendazole sulfoxide (ABZ-SO, 1 mg/mL), were prepared in DMSO. The stock solutions of the benzimidazole compounds were serially diluted in Dulbecco’s Modified Eagle’s Medium (DMEM) to prepare working solutions in the range of 0.28 to 154 µM. For ABZ-SO, working solutions in DMEM were prepared in the range of 0.03 to 2.17 µM. 0.25% DMSO in DMEM was used as the negative control.
The assays were performed in triplicate in 24-well flat-bottom cell culture microplates (NUNC, Denmark) containing 2 mL of the working solutions of each compound or 0.25% DMSO. Ten cysts were then placed into each well and microplates were incubated at 37°C in a humidified atmosphere (98% relative humidity) under 5% CO2 for 11 d. The culture medium was changed every two days. The parasites were observed every day and the integrity, motility, morphology and mortality were evaluated using an inverted light microscope (ICM 405; Carl Zeiss, U.S.A.). On day 12, the mortality of parasites was established using the Trypan Blue exclusion test.9) For each compound, the effective concentration required to kill 50% of the cysts (EC50) was calculated according to the Hill’s equation using the GraphPad software (Prism, version 8 for Windows, U.S.A.).
In Silico Toxicological EvaluationThe structures of the synthesized compounds (1–3), ABZ and ABZ-SO were built using Avogadro10) molecular software and uploaded to DataWarrior Osiris 4.4.4 program (Actelion Pharmaceuticals) to predict the molecular and toxicological properties in terms of druglikeness, c Log P, polar surface area (PSA) value, mutagenicity, tumorigenicity, irritability and reproductive effectiveness.
Molecular Modeling Studies
Molecular DockingProtein PreparationThe MOL2 file of the optimized Gallus gallus (G. gallus) β-tubulin (PDB: 5CA1)11) with the protonated state of E198 residue was generated with the OpenBabel toolbox.12)
Ligand PreparationThe 3D structures of the 1,5- and 1,6-tautomers of compounds 1–3, ABZ, and ABZ-SO enantiomers were energy-minimized with the obminimize tool of the OpenBabel toolbox using the GAFF force field.
DockingMolecular docking of compounds 1–3, ABZ, and ABZ-SO enantiomers in the nocodazole (NZ) binding site of the G. gallus β-tubulin was performed with the rDock software13) using previously described methodology.14) For this study, we carried out 500 runs of both benzimidazole tautomers of each compound with the rbdock tool. The intermolecular scores and ligand root mean square deviation (RMSD) values were retrieved with the sdreport and sdrmsd tools of the same software, respectively. The figures were generated with Gnuplot 5.015) and PyMOL v0.9.16)
Molecular DynamicsThe docked β-tubulin-compound complexes were evaluated through 50 ns of MD simulations using the GROMACS 5.1.4 package17) and the AMBER99SB-ILDN force field.18) The topology and atom types of the compounds were parametrized with the ACPYPE interface19) in the amber framework, calculating the partial charges using the Gasteiger method. The complexes were solvated with a TIP3P water model and neutralized employing sodium and chloride ions. The systems were submitted to energy-minimization using 5000 steps of the steepest-descent and equilibrated for 5 ns in a canonical ensemble using the Berendsen thermostat.20) Further equilibration in an isothermal-isobaric ensemble for 5 ns at 1 bar was performed using the Parrinello–Rahman barostat.21) The Poison–Boltzmann Surface Area (MM-PBSA) binding free energies were calculated with the g_mmpbsa.py script.22)
In Vivo ActivityCompound 3 showed the best in vitro cysticidal activity and good toxicological characteristics; therefore, its in vivo cysticidal activity and pharmacokinetic properties were further evaluated. For the in vivo cysticidal activity, the T. crassiceps murine cysticercosis model was used.23) The selected dose was 70 mg/kg, which represents an equimolar dose with respect to the effective dose of ABZ (50 mg/kg) previously reported.23) Solid dispersions of 14 mg/mL of compound 3 or 10 mg/mL of ABZ (positive control) in 0.5% carboxymethyl cellulose (CMC) were prepared; 0.5% CMC was used as the negative control. All dispersions were stored at −5°C for a maximum period of 5 d.
Thirty female BALB/C mice (16–18 g, Harlan Laboratories, Mexico City, Mexico) were infected by an intraperitoneal injection of 30 T. crassiceps cysts (ORF strain) previously suspended in 0.5 mL of sterile 0.9% saline solution. Ten days after infection, mice were randomly allocated to three experimental groups (10 mice per group) as follows: (1) Negative control group, which received 0.5% CMC solution, (2) compound 3 group, and (3) ABZ group (positive control). Each mouse received 0.1 mL of the corresponding treatment by intragastric administration on 20 consecutive days. On day 21, mice were sacrificed by cervical dislocation before the cysts were removed from the peritoneal cavity and counted to calculate the cysticidal efficacy. The cysticidal efficacy was expressed as the percentage of parasites recovered from the treated mice with respect to that obtained from the negative control mice.
A one-way ANOVA followed by Dunnett’s test was conducted between the negative control group and the different treatments. Statistical analysis was carried out using SPSS software (V. 15.0) and differences were considered significant at p < 0.05.
Microscopic EvaluationTo observe the effect of compound 3 on the parasite tissue, living cysts were recovered from the peritoneum of mice after 20 d of treatment. Cysts were washed with 0.9% saline solution and observed using an ICM 405 microscope (Carl Zeiss, U.S.A.). For transmission electronic microscopy (TEM), cysts were fixed with 2.5% glutaraldehyde (1 h), rinsed with 0.1 M phosphate buffered saline solution (pH 7.4), and post-fixed in 0.5% osmium tetroxide before being dehydrated in a graded ethanol series and embedded in EPON resin. Subsequently, the resin was polymerized at 60°C overnight and ultra-thin sections (thickness 60 nm) were cut using an ultramicrotome (Leica EM UC6). Sections were recovered over covered Formvar copper grids, stained with 3% uranyl acetate/0.3% lead citrate and examined using a JEOL 1011 transmission electron microscope at 70 kV. TEM images were obtained and scale bars, lettering and arrows were added using Adobe Photoshop CS3 Extended (Version 10.0).
Pharmacokinetic StudyTwenty male Wistar rats (300–350 g, Instituto Nacional de Neurología y Neurocirugía, Mexico City, Mexico) were used for the study. Prior to the experiments, rats were fasted for at least 12 h. Each animal was anesthetized with inhaled isoflurane (1.5–2% isoflurane in 100% oxygen). After the animal was anesthetized, a catheter was implanted into the tail vein for blood collection according to the procedure previously described by de Jong et al.24) Briefly, the vein was exposed after making an incision 3 cm from the tail base. A previously heparinized polyethylene cannula (inner diameter 0.058 cm and outer diameter 0.097 cm) (Becton Dickinson, Franklin Lakes, NJ, U.S.A.) was introduced into the vein and fixed with a 2–0 surgical suture. Rats were maintained for a period of 1–2 h to allow complete recovery before the pharmacokinetic study was started.
The rats were administered a single oral dose of 21 mg/kg of compound 3, and blood samples (approximately 700 µL, 4 samples per animal) were collected into heparinized tubes at 0, 0.5, 1.5, 3, 5, 6, 7, 9, 12, 24 and 30 h after drug administration (5 replicates per sampling time). Blood samples were centrifuged at 3000 rpm for 15 min to obtain plasma, which was stored at −70°C prior to LC-MS/MS analysis.
Plasma concentration–time data were analyzed using Phoenix WinNonLin 6.4 by a non-compartmental analysis model (Pharsight Corp., Mountain View, CA, U.S.A.). Peak plasma concentration (Cmax), time to reach peak plasma concentration (Tmax), area under the curve to the last measurable plasma concentration point (AUC0 → t), elimination half-life (t1/2) and mean residence time (MRT) were calculated.
Analytical AssayCompound 3 plasma levels were determined by LC-MS/MS. The system consisted of an Agilent 1100 HPLC (quaternary pump and autosampler) (Palo Alto, CA, U.S.A.) coupled to an ABSciex 3200 QTrap linear ion trap quadrupole mass spectrometer (ABSciex, Darmstadt, Germany) with an electrospray ionization (ESI) interface, which was operated in the positive ionization mode. Data acquisition and processing were performed using the Analyst software 1.6.2 version (Applied Biosystems, Foster City, CA, U.S.A.), and quantitative analysis was performed using MultiQuant software version 3.0 (Applied Biosystems).
The chromatographic separation was carried out on a Gemini C18 analytical column (150 × 4.6 mm, 5 µm, Phenomenex, Torrance, CA, U.S.A.) attached to a pre-column (Phenomenex C18 ODS). The mobile phase was composed of formic acid 5 mM–acetonitrile–methanol (30 : 33 : 37, v/v/v) at a flow rate of 0.8 mL/min. Mass spectrometer optimized conditions were: curtain gas 10 psi, ion spray voltage 5500 V, temperature 600°C, nebulization gas 40 psi and heating gas 40 psi. Quantification was performed using multiple reaction monitoring (MRM) of the following transitions: m/z 370 → 337 for compound 3, and m/z 282 → 240 for ABZ-SO as the internal standard (IS).
Extraction ProcedurePlasma (300 µL) was transferred to an assay tube and spiked with 100 µL of IS (0.3 µg/mL). Methyl-tert-butyl ether (5 mL) was added and samples were vortex-shaken for 5 min. After centrifugation at 3000 rpm for 20 min, samples were frozen at −70°C for 20 min and the organic upper phase was transferred to a clean tube. Samples were evaporated to dryness at 45°C under a nitrogen stream. Finally, samples were reconstituted with 50 µL of the mobile phase (5 mM formic acid–acetonitrile–methanol 30 : 33 : 37, v/v/v), and 10 µL was injected into the chromatographic system. The limit of detection of the method was 1.5 × 10−3 µg/mL and the assay range was 1.5 × 10−3–0.1 µg/mL.
The regression equation was y = 0.023x + 0.0035 with a correlation coefficient (r2) of 0.9977. The precision of the intra-day assay was between 3.26 and 5.70%, while for the inter-day assay precision was between 3.20 and 5.19%. In terms of accuracy, the deviation with respect to the nominal concentrations ranged from 5.68 to 7.91% for the intra-day assays and from 5.23 to 8.04% for the inter-day assays.
Results and Discussion
ChemistryCompounds (1–3) were synthesized according to the sequence of reactions outlined in Chart 1. The synthesis began with 3,4-diaminobenzoic acid, which was reacted with di-tert-butyl dicarbonate to form 3,4-bis[(tert-butoxycarbonyl) amino] benzoic acid (6). Subsequently, this compound was activated with CDI using acetonitrile as a solvent. Following formation of the intermediate [4-(1H-imidazol-1-ylcarbonyl)-1,2-phenylene] bis tert-butyl carbamate, the proper amine was added to generate the amide intermediates (7–9). Later, the N-BOC protecting groups were removed with trifluoroacetic acid to yield the benzamides (10–12), which were used immediately for the next reaction. Finally, target compounds 1–3 were generated by cyclocondensation of the corresponding benzamides with 1,3-dimethoxycarbonyl-S-methyl-isothiourea. The structure of the new molecules was confirmed by IR, 1H-NMR and MS. All compounds were obtained with yields ranging from 50 to 65.4%.
In Vitro ActivityResults showed that compounds 1–3 exhibited activity against T. crassiceps cysts (ORF strain) and the effect was concentration-time dependent.25) Cysts showed a gradual loss of cystic fluid with the subsequent changes in shape and size; furthermore, their motility was decreased. EC50 values showed that compounds 1 and 2 had a lower potency (8.32 µM [6.62–10.62] and 4.12 µM [3.36–6.52], respectively) than ABZ-SO (0.52 µM [0.46–0.62]). Interestingly, the potency of compound 3 (EC50, 0.86 µM [0.31–1.07]) was similar to that of the positive control.
In Silico Toxicity StudyData obtained from DataWarrior Osiris26) are presented in Table 1. Physicochemical data revealed that the molecular properties of the benzimidazole derivatives fulfilled Lipinski’s rule of five (RO5), which is commonly used to predict druglikeness and oral bioavailability.27) The molecular weight of the three compounds was less than 500, and the partition coefficient (Log P) values were less than 5, suggesting a good permeability across cell membranes. With respect to the number of hydrogen bond donors and acceptors, only derivative 2 was not in accordance with Lipinski’s RO5, with more than 10 hydrogen bond acceptors. Besides hydrogen bonds, Veber et al. have suggested a PSA ≤ 140 Å2 as a criterion for oral bioavailability; compound 2 was found to have a PSA value greater than this limit suggesting a poor permeability across membranes.28) In silico toxicity risk information generated by the program suggests a low mutagenic, tumorigenic and irritant potential of the compounds 1–3.
Table 1. Prediction of Molecular Properties of Compounds
1–
3, Toxicity and Drug-Likeness in Datawarrior-Osiris
| Compound | Physicochemical properties | Toxicity risks |
|---|
| MWb) | c Log Pc) | c Log Sd) | HBAe) | HBDf) | TSAg) | Relative PSAh) | Polar surface area | Drug likeness | Mutagenic | Tumorigenic | Reproductive effective | Irritant |
|---|
| 1 | 382.37 | 2.305 | −4.224 | 9 | 3 | 290.4 | 0.3684 | 122.4 | −3.2009 | − | − | + | − |
| 2 | 370.32 | 2.040 | −4.337 | 11 | 4 | 271.7 | 0.4630 | 154 | −6.377 | − | − | + | − |
| 3 | 369.33 | 1.300 | −4.543 | 10 | 3 | 274.0 | 0.4173 | 141.9 | −6.9818 | − | − | + | − |
| ABZa) | 265.33 | 2.964 | −4.105 | 5 | 2 | 204.9 | 0.3754 | 92.31 | −2.9435 | + | − | + | − |
| ABZ-SOa) | 281.33 | 1.992 | −2.848 | 6 | 2 | 210.1 | 0.3910 | 103.3 | −4.7295 | − | − | + | − |
a) Positive control, b) Molecular weight, c) Calculated partition coefficient, d) Calculated aqueous solubility, e) Number of hydrogen bond acceptors, f) Number of hydrogen bond donors, g) Total surface area, h) Polar surface area. Note: (−), no risk; (+), high risk.
To explore the possible binding modes of compounds 1–3 to β-tubulin and to explain the activity differences, we docked the benzimidazole derivatives into the NZ binding site of β-tubulin.11) To our knowledge, the complete β-tubulin sequence of T. crassiceps has not been published; however, residues that constitute the NZ binding site are highly conserved in β-tubulin sequences of other Taenia species (e.g., T. asiatica, UniProtKB A0A0R3VWM0).29) Considering that this protein is well-conserved between species, we employed the crystal structure of the G. gallus β-tubulin complexed with NZ (PDB: 5CA1) to perform our molecular docking studies.11)
Based on our recent study, we docked the benzimidazole derivatives in the G. gallus β-tubulin taking into account the protonated state of E198, the presence of a structural water molecule located between G235 and C239 and two benzimidazole tautomers (1,5/6).14) These considerations have proved to be key structural features required for the binding of other tubulin polymerization inhibitors to this binding site.30) Figure 2A shows the rDock score profile of the predicted binding modes and its RMSD when compared with the lowest binding free energy conformation. Contrary to what is observed in the NZ and ABZ-SO binding modes, the best rDock scores with the lowest RMSD values were obtained with the 1,5-tautomer due to the presence of the bulky substituent (Fig. 2B). It should be mentioned that the 1,5-tautomer binding mode is also favored for ABZ. Despite these differences in binding modes, benzimidazole derivatives retain the hydrogen bond interactions with amino acids N165, E198 and V236 that are observed with NZ. Furthermore, compound 3 had the highest probability of binding to the 1,5-tautomer and the lowest binding free energy score, followed by compounds 2 and 1, respectively.
Additionally, the benzimidazole-2-carbamate derivatives complexed with the G. gallus β-tubulin were submitted to 50 ns of MD simulations to evaluate their stability in the binding site (Fig. 2C). Interestingly, the simulation revealed low ligand position RMSD values for all the compounds, suggesting high stability at the binding site. Moreover, the calculated MM-PBSA binding free energies of the complexes showed the following affinity order: 3 > 1 > 2, being the hydrophobic protein-ligand interactions with V236, T237, L240, L246 and L253 those with lower energy contributions to the binding (Fig. 2D).
Based on the in vitro and docking results, compound 3 was selected for further in vivo evaluation.
In Vivo Cysticidal ActivityResults of the in vivo evaluation showed that compound 3 was well-tolerated by the animals, with no abnormalities in behavior, food or water consumption and general activity. The cysticidal efficacy based on parasite reduction relative to the negative control was 50.39% (p < 0.05), with no significant difference with respect to the positive control (ABZ, 47.16%) (Fig. 3). After the administration of compound 3, we found that, as with ABZ, the recovered cysts were smaller than those found in the negative control group (0.5% CMC). Moreover, cysts lost their ruggedness as well as their characteristic movements.
Microscopic Evaluation of in Vivo Recovered CystsUltrastructural observations showed that all cyst structures remained unaltered in the negative control group (Figs. 4 a–c). In contrast, treatment with the compound 3 affected all structures in the parasite tissue. A significant reduction in the length and number of microtriches (Fig. 4d) was found in the tegument. In the germinal layer, the main effect was on muscular fibers and flame cells, in which partial fragmentation was observed (Figs. 4e–f). These effects are similar to those previously reported for ABZ, which suggests that compound 3 shares the same mechanism of action.31,32)
Pharmacokinetic StudyFigure 5 shows the pharmacokinetic profile of compound 3 after an oral administration. The sensitivity of the method allowed us to quantify plasma concentrations up to 30 h post-dose. The pharmacokinetic parameters are shown in Table 2. Although plasma levels were low, the half-life and MRT were long. The maximum plasma concentrations were observed at 7 h, indicating a slow absorption process. The plasma concentration–time profile showed two peaks, which could be attributed to the presence of biliary salts that increase the solubility of residual compound in the small intestine. As observed with other benzimidazoles, enterohepatic recirculation could also be an explanation.33,34) Further studies should be performed to elucidate the reasons for these observations.
Table 2. Pharmacokinetic Parameters of Compound
3 in Rat after an Oral Administration of 21 mg/kg (
n = 5)
| Cmax (ng/mL) | tmax (h) | t1/2 (h) | AUC(0 → t) (h*ng/mL) | MRT(0 → t) (h) |
|---|
| Mean | 50.10 | 6.8 | 6.06 | 660.34 | 11.93 |
| (S.D.) | (17.29) | (0.45) | (0.87) | (274.23) | (0.90) |