Experimental
GeneralThe following instruments were used to obtain physical data: Agilent NMR system (1H-NMR: 600 MHz, 13C-NMR: 150 MHz) spectrometer for 1H- and 13C-NMR data using tetramethylsilane or solvent residual peak (CHCl3: δ 7.26) as internal standards; a JASCO FT/IR-5300 IR spectrometer for IR spectra; a JEOL JMS-S3000 mass spectrometer for matrix-assisted laser desorption/ionization-time-of-flight (MALDI-TOF) MS; and a Waters Q-Tof Ultima API mass spectrometer for electrospray ionization (ESI)-TOF MS. Silica gel (Kanto, 40–100 µm) and precoated TLC plates (Merck, 60F254) were used for column chromatography and TLC. Spots on TLC plates were detected by spraying acidic p-anisaldehyde solution (p-anisaldehyde: 25 mL, c-H2SO4: 25 mL, AcOH: 5 mL, EtOH: 425 mL) or phosphomolybdic acid solution (phosphomolybdic acid: 25 g, EtOH: 500 mL) with subsequent heating. Cell culture and assay for growth–inhibitory activity were performed with a previously reported method.6–8)
Unless otherwise noted, the entire reaction was performed under a N2 atmosphere. After workup, the organic layer was dried over Na2SO4. Perfect separation of geometrical isomers of biakamide analog at vinyl chloride moiety proved difficult. Therefore the yields of the respective reactions are shown as a mixture of geometrical isomers. The parts of purified compound were used in the assay for growth–inhibitory activity and measurement of physical data.
All compounds used in the assay for growth–inhibitory activity of cancer cells were finally purified by HPLC (Cosmosil 5C18-MS-II, 10 mm i.d. × 250 mm).
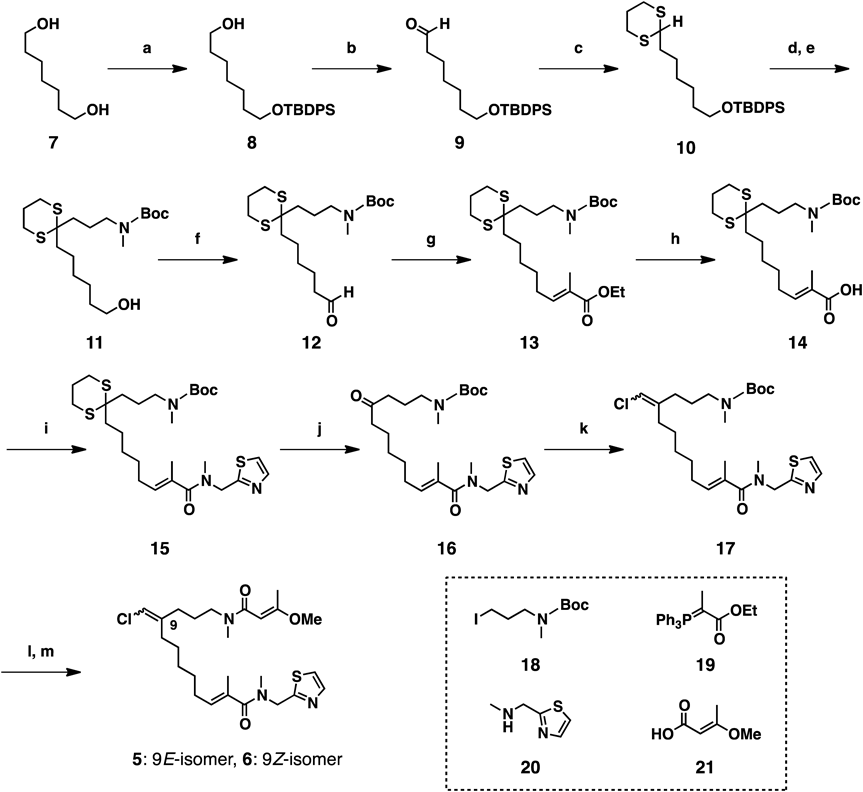
Chemistry7-((tert-Butyldiphenylsilyl)oxy)heptan-1-ol (8)Et3N (1.6 mL, 11 mmol, 1.2 equiv.) and tert-butylchlorodiphenylsilane (TBDPSCl, 2.5 mL, 11 mmol, 1.2 equiv.) were added to a solution of 7 (1.24 g, 9.4 mmol) in CH3CN : hexane (1 : 3, 28 mL), and the mixture was stirred at room temperature (r.t.) for 8 h. The reaction was quenched by addition of sat. NaHCO3 aq., and the whole mixture was extracted with AcOEt. Concentration of the AcOEt extract in vacuo gave a crude product which was purified by SiO2 column chromatography (hexane : AcOEt = 5 : 1 to 1 : 1) to give 8 (1.92 g, 60%) as a colorless oil. The spectroscopic and physical data were identical to those reported.9)
7-((tert-Butyldiphenylsilyl)oxy)heptanal (9)Iodobenzene diacetate (1.75 g, 5.4 mmol, 1.2 equiv.) and TEMPO (73.8 mg, 0.47 mmol, 0.1 equiv.) were added to a solution of 8 (1.75 g, 4.7 mmol) in CH2Cl2 (30 mL), and the mixture was stirred for 3.5 h at r.t. The reaction was quenched by the addition of Na2S2O3/NaHCO3 aq., and the whole mixture was extracted with AcOEt. The AcOEt extract was successively washed with sat. NH4Cl aq., NaHCO3 aq., and brine. Concentration of the AcOEt extract in vacuo gave a crude product which was purified by SiO2 column chromatography (hexane : AcOEt = 6 : 1) to give 9 (1.59 g, 91%) as a colorless oil. The spectroscopic and physical data were identical to those reported.9)
((6-(1,3-Dithian-2-yl)hexyl)oxy)(tert-butyl)diphenylsilane (10)1,3-Propanedithiol (89 µL, 0.87 mmol, 1.3 equiv.) and iodine (17 mg, 0.067 mmol, 0.10 equiv.) were added to a solution of 9 (226 mg, 0.57 mmol) in CHCl3 (10 mL), and the mixture was stirred at r.t. for 0.5 h. The reaction was quenched by the addition of Na2S2O3/NaHCO3 aq., and the whole mixture was extracted with AcOEt. The AcOEt extract was washed with Na2S2O3/NaHCO3 aq. and brine. Concentration of the AcOEt extract in vacuo gave a crude produt, which was purified by SiO2 column chromatography (Hexane : AcOEt = 50 : 1) to give 10 (311 mg, quant.) as a colorless oil.
1H-NMR (600 MHz, CDCl3) δ: 1.05 (9H, s), 1.13–1.43 (4H, m), 1.43–1.66 (4H, m), 1.73 (2H, dd, J = 15.5, 7.1 Hz), 1.86 (1H, ddd, J = 14.3, 11.6, 8.3 Hz), 2.05–2.20 (1H, m), 2.77–2.93 (4H, m), 3.65 (2H, t, J = 6.5 Hz), 4.04 (1H, t, J = 6.9 Hz), 7.40 (6H, m), 7.67 (4H, d, J = 6.7 Hz). 13C-NMR (150 MHz, CDCl3) δ: 19.4, 25.7, 26.2, 26.8, 27.0, 29.1, 30.6, 30.7, 32.6, 35.6, 47.8, 64.0, 127.7 (4C), 129.6 (2C), 134.2 (2C), 135.7 (4C). IR (KBr) cm−1: 3070, 2931, 1427, 1109. MS (ESI-TOF) m/z: 481 [M + Na]+. High resolution (HR)-MS (ESI-TOF) m/z: 481.2031 calcd for C26H38OSiS2Na; Found: 481.2020.
tert-Butyl (3-(2-(6-Hydroxyhexyl)-1,3-dithian-2-yl)propyl)(methyl)carbamate (11)n-BuLi (1.59 M in n-hexane, 1.4 mL, 2.3 mmol, 1.5 equiv.) was added to a solution of 10 (745 mg, 1.5 mmol) in anhydrous tetrahydrofuran (THF, 20 mL), and the mixture was stirred for 20 min. A solution of 18 (710 mg, 2.4 mmol, 1.6 equiv.) in anhydrous THF (5 mL) was added dropwise to the organolithium solution via cannula at r.t. over 10 min, and the resulting mixture was stirred at r.t. for 0.5 h. Then, the reaction was quenched by the addition of H2O, and the whole mixture was extracted with AcOEt. The AcOEt extract was washed with brine. Concentration of the AcOEt extract in vacuo, the residue was dissolved in THF (10 mL). TBAF solution (1 M in THF, 2.6 mL) was added to the solution at r.t., and the mixture was stirred at r.t. for 8 h. The reaction was quenched by the addition of sat. NH4Cl aq., and the whole mixture was extracted with AcOEt, and the AcOEt extract was washed with sat. NH4Cl aq. and brine. Concentration of the AcOEt extract in vacuo gave a crude product, which was purified by SiO2 column chromatography (Hexane : AcOEt = 3 : 1) to give 11 (533 mg, 69%, 2 steps) as a colorless oil.
1H-NMR (600 MHz, CDCl3) δ: 1.02–1.36 (15H, m), 1.40 (2H, br s), 1.48 (2H, br s), 1.68 (4H, br s), 1.78 (2H, br s), 2.63 (4H, br s), 2.69 (3H, br s), 2.89 (1H, s), 3.07 (2H, br s), 3.44 (2H, dd, J = 11.5, 4.9 Hz). 13C-NMR (150 MHz, CDCl3) δ: 22.2, 22.6, 23.6, 23.7, 25.2, 25.4, 25.7, 28.2, 29.3, 32.4, 33.9, 34.5, 34.7, 38.0, 47.8, 48.6, 52.7, 62.1, 79.1, 155.6. IR (KBr) cm−1: 3442 (br), 2935, 1679, 1397, 1153, 756. MS (ESI-TOF) m/z: 414 [M + Na]+. HR-MS (ESI-TOF) m/z: 414.2113 calcd for C19H37NO3S2Na; Found: 414.2113.
tert-Butyl Methyl(3-(2-(6-oxohexyl)-1,3-dithian-2-yl)propyl)carbamate (12)Molecular sieves 4A (100 mg) and N-methylmorpholine N-oxide (NMO, 47 mg, 0.40 mmol, 1.5 equiv.) were added to a solution of 11 (113 mg, 0.27 mmol) in CH2Cl2 (4 mL), and the mixture was stirred at r.t. for 20 min. Then, TPAP (4.7 mg, 0.013 mmol, 0.050 equiv.) was added to the solution, and the resulting mixture was stirred at r.t. for 40 min. The reaction was quenched by the addition of Na2S2O3/NaHCO3 aq., and the whole mixture was extracted with AcOEt. The AcOEt extract was washed with sat. NH4Cl aq. and brine. Concentration of the AcOEt extract in vacuo gave a crude product which was purified by SiO2 column chromatography (hexane : AcOEt = 5 : 1) to give 12 (78 mg, 70%) as a colorless oil.
1H-NMR (600 MHz, CDCl3) δ: 1.28 (2H, m), 1.38 (11H, m), 1.57 (4H, m), 1.76 (4H, br s), 1.86 (2H, br), 2.37 (2H, t, J = 7.2 Hz), 2.74 (7H, m), 3.15 (2H, br s), 9.69 (1H, s). 13C-NMR (150 MHz, CDCl3) δ: 11.9, 13.5, 13.8, 15.1, 20.1, 22.3, 23.2, 24.9, 25.4, 27.9, 28.0, 28.9, 33.5, 34.5, 37.8, 47.4, 48.2, 52.3, 59.7, 60.3, 78.4, 127.2, 141.3, 155.0, 167.2. IR (KBr) cm−1: 2934, 1692, 1393, 1148. MS (ESI-TOF) m/z: 412 [M + Na]+. HR-MS (ESI-TOF) m/z: 412.1956 calcd for C19H35NO3S2Na; Found: 412.1938.
Ethyl (E)-8-(2-(3-((tert-Butoxycarbonyl)(methyl)amino)propyl)-1,3-dithian-2-yl)-2-methyloct-2-enoate (13)Methyl 2-(triphenyl-λ5-phosphanylidene)acetate (19, 298 mg, 0.82 mmol, 3.0 equiv.) was added to a solution of 12 (107 mg, 0.27 mmol) in toluene (8 mL), and the mixture stirred at 100°C for 10 h. Then, the reaction was quenched by the addition of sat.NH4Cl aq. The whole mixture was extracted with AcOEt, and the AcOEt extract was washed with NH4Cl aq. and brine. Concentration of the AcOEt extract in vacuo gave a crude product which was purified by SiO2 column chromatography (hexane : AcOEt = 4 : 1) to give 13 (128 mg, 99%) as a colorless oil.
1H-NMR (600 MHz, CDCl3) δ: 0.85–1.09 (5H, m), 1.17 (11H, m), 1.36 (2H, br s), 1.45–1.72 (9H, m), 1.90 (2H, br s), 2.42–2.60 (7H, m), 2.95 (2H, br s), 3.77–3.96 (2H, dd, J = 15.5, 6.5 Hz), 6.45 (1H, br s). 13C-NMR (150 MHz, CDCl3) δ: 11.8, 13.5, 13.8, 15.1, 20.1, 22.3, 23.3, 24.9, 25.4, 27.1, 27.9, 28.0, 28.7, 28.8, 28.9, 33.5, 34.5, 37.8, 47.5, 48.2, 52.3, 59.3, 59.7, 60.3, 78.4, 127.2, 132.6, 141.0, 141.3, 142.2, 155.0, 167.2. IR (KBr) cm−1: 2935, 1698, 1393, 1274, 1150. MS (ESI-TOF) m/z: 496 [M + Na]+. HR-MS (ESI-TOF) m/z: 496.2531 calcd for C24H43NO4S2Na; Found: 496.2554.
(E)-8-(2-(3-((tert-Butoxycarbonyl)(methyl)amino)propyl)-1,3-dithian-2-yl)-2-methyloct-2-enoic Acid (14)LiOH (23 mg, 0.95 mmol, 5.0 equiv.) was added to a solution of 13 (90 mg, 0.19 mmol) in THF : MeOH : H2O (1 : 1 : 1, 6 mL), and the mixture was stirred at r.t. for 13 h. The reaction was quenched by the addition of 5% HCl, and the whole mixture was extracted with AcOEt. Concentration of the AcOEt extract in vacuo gave a crude product which was purified by SiO2 column chromatography (hexane : AcOEt = 1 : 1) to give 14 (84 mg, 99%) as a colorless oil.
1H-NMR (600 MHz, CDCl3) δ: 1.24–1.33 (2H, m), 1.33–1.48 (13H, m), 1.60 (2H, dt, J = 14.9, 7.1 Hz), 1.76–1.83 (7H, m), 1.89 (2H, br s), 2.16 (2H, dd, J = 14.7, 7.3 Hz), 2.64–2.87 (7H, m), 3.19 (2H, br s), 6.84 (1H, t, J = 7.3 Hz), 10.22 (1H, br). 13C-NMR (150 MHz, CDCl3) δ: 12.1, 22.7, 23.9, 25.4, 26.0, 28.3, 28.5, 28.8, 29.5, 34.2, 35.0, 38.4, 48.6, 53.0, 79.4, 127.2, 144.8, 155.9, 173.4. IR (KBr) cm−1: 2933, 1691, 1394, 1152. MS (ESI-TOF) m/z: 468 [M + Na]+. HR-MS (ESI-TOF) m/z: 468.2218 calcd for C22H39NO4S2Na; Found: 468.2197.
tert-Butyl (E)-Methyl(3-(2-(7-methyl-8-(methyl(thiazol-2-ylmethyl)amino)-8-oxooct-6-en-1-yl)-1,3-dithian-2-yl)propyl)carbamate (15)1-Hydroxybenzotriazole (HOBt, 36 mg, 0.26 mmol, 1.4 equiv.), N-methyl-1-(thiazol-2-yl)methanamine (20, 36 mg, 0.28 mmol, 1.5 equiv.), Et3N (150 µL, 1.1 mmol, 4.0 equiv.), and 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDCI·HCl, 51 mg, 0.26 mmol, 1.4 equiv.) were added to a solution of 14 (84 mg, 0.19 mmol) in CH2Cl2 (5 mL), and the mixture was stirred at r.t. for 10 h. Then, the reaction was quenched by the addition of sat. NH4Cl aq. The whole mixture was extracted with AcOEt, and the AcOEt extract was washed with sat. NH4Cl aq., NaHCO3 aq., and brine. Concentration of the AcOEt extract in vacuo gave a crude product which was purified by SiO2 column chromatography (hexane : AcOEt = 1 : 2) to give 15 (82 mg, 78%) as a colorless oil.
1H-NMR (600 MHz, CDCl3) δ: 1.06–1.41 (15H, m), 1.46–1.58 (2H, m), 1.59–1.87 (9H, m), 1.94–2.05 (2H, m), 2.56–2.79 (7H, m), 2.91 (3H, s), 3.09 (2H, br s), 4.75 (2H, s), 5.51 (1H, br s), 7.22 (1H, br s), 7.59 (1H, br s). 13C-NMR (150 MHz, CDCl3) δ: 14.0, 19.9, 22.3, 22.6, 23.6, 25.2, 25.8, 27.4, 28.3, 28.5, 28.8, 29.3, 31.9, 33.1, 33.9, 34.6, 34.8, 35.4, 36.8, 38.1, 47.4, 47.8, 48.2, 48.6, 52.7, 52.8, 79.0, 119.9, 120.1, 129.5, 130.6, 131.9, 141.9, 142.0, 142.7, 155.5, 166.1, 166.6, 172.0, 173.5. IR (KBr) cm−1: 3019, 1682, 1216, 909, 756. MS (ESI-TOF) m/z: 578 [M + Na]+. HR-MS (ESI-TOF) m/z: 578.2521 calcd for C27H45N3O3S3Na; Found: 578.2543.
tert-Butyl (E)-Methyl(11-methyl-12-(methyl(thiazol-2-ylmethyl)amino)-4,12-dioxododec-10-en-1-yl)carbamate (16)Iodine (149 mg, 0.59 mmol, 4.0 equiv.) was added to a solution of 15 (82 mg, 0.15 mmol) in a mixture of CH3CN (4 mL) and sat. NaHCO3 aq. (2 mL) at 0°C, and the mixture was stirred at 0°C for 0.5 h. The reaction was quenched by the addition of Na2S2O3/NaHCO3 aq., and the whole mixture was extracted with AcOEt. Then, the AcOEt extract was washed with Na2S2O3/NaHCO3 aq. and brine. Concentration of the the AcOEt extract in vacuo gave a crude product which was purified by SiO2 column chromatography (hexane : AcOEt = 1 : 3) to give 16 (61 mg, 89%) as a colorless oil.
1H-NMR (600 MHz, CDCl3) δ: 1.20 (2H, br s), 1.29–1.42 (11H, m), 1.49 (2H, br s), 1.66–1.76 (2H, m), 1.81 (3H, s), 2.04 (2H, br d, J = 7.1 Hz), 2.32 (4H, t, J = 7.0 Hz), 2.76 (3H, s), 2.97 (3H, s), 3.14 (2H, br s), 4.81 (2H, s), 5.57 (1H, br s), 7.28 (1H, d, J = 2.8 Hz), 7.66 (1H, br s).13C-NMR (150 MHz, CDCl3) δ: 14.2, 20.1, 21.5, 21.9, 23.5, 27.4, 28.5, 28.7, 28.9, 29.2, 29.7, 34.0, 35.6, 37.0, 39.4, 39.5, 42. 7, 47.6, 48.1, 48.4, 51.8, 79.2, 79.3, 120.1, 120.3, 129.6, 130.8, 132.1, 142.1, 142.9, 155.8, 166.3, 166.9, 172.2, 173.0, 210.1, 210.4. IR (KBr) cm−1: 2931, 1692, 1394, 1170. MS (ESI-TOF) m/z: 488 [M + Na]+. HR-MS (ESI-TOF) m/z: 488.2559 calcd for C24H39N3O4SNa; Found: 488.2559.
tert-Butyl ((4EZ,10E)-4-(Chloromethylene)-11-methyl-12-(methyl(thiazol-2-ylmethyl)amino)-12-oxododec-10-en-1-yl)(methyl)carbamate (17)Lithium bis(trimethylsilyl)amide (LHMDS) solution (1.0 M in THF, 515 µL, 0.53 mmol, 5.0 equiv.) was added to a solution of (chloromethyl)triphenylphosphonium chloride (185 mg, 0.51 mmol, 5.0 equiv.) in anhydrous THF (3 mL), and the mixture was stirred at r.t. for 10 min. A solution of 16 (50 mg, 0.11 mmol) in anhydrous THF (1 mL) was added dropwise to the ylide solution via cannula at r.t. over 5 min, and the resulting mixture was stirred at r.t. for 1 h. Then, the reaction was quenched by the addition of sat. NH4Cl aq. The whole mixture was extracted with AcOEt, and the AcOEt extract was washed with NH4Cl aq. and brine. Concentration of the AcOEt extract in vacuo gave a crude product which was purified by SiO2 column chromatography (hexane : AcOEt = 1 : 2) to give 17 (57 mg, quant.) as a colorless oil.
1H-NMR (600 MHz, CDCl3) δ: 1.21–1.48 (15H, m), 1.56–1.66 (2H, m), 1.86 (3H, s), 1.97–2.03 (2H, t, J = 8.0 Hz), 2.06–2.23 (4H, m), 2.77–2.88 (3H, m), 3.04 (3H, s), 3.19 (2H, br s), 4.89 (2H, s), 5.65 (1H, br s), 5.72–5.81 (1H, m), 7.34 (1H, d, J = 3.3 Hz), 7.73 (1H, d, J = 3.2 Hz). IR (KBr) cm−1: 3020, 1216, 771. MS (ESI-TOF) m/z: 520 [M + Na]+. HR-MS (ESI-TOF) m/z: 520.2377 calcd for C25H40N3O3S35ClNa; Found: 520.2368.
14,15-Dinor-biakamide C, D (5, 6)Trifluoroacetic acid (TFA, 255 µL, 3.4 mmol, 30 equiv.) was added to a solution of 17 (57 mg, 0.11 mmol) in CH2Cl2 (2 mL), and the mixture was stirred for 1 h. Then, the reaction was quenched by the addition of sat. NaHCO3 aq. The whole mixture was extracted with AcOEt, and the AcOEt extract was washed with sat. NaHCO3 aq. and brine. Concentration of the AcOEt extract in vacuo gave an amine, which was used in the next reaction without further purification. The above crude product was dissolved to CH2Cl2 (2 mL). HOBt (14 mg, 0.11 mmol, 1 equiv.), 21 (12 mg, 0.11 mmol, 1 equiv.), Et3N (90 µL, 0.65 mmol, 6.0 equiv.), and EDCI·HCl (20 mg, 0.11 mmol, 1.0 equiv.) were added to the solution, and the resulting mixture was stirred at r.t. for 15 h. Then, the reaction was quenched by the addition of sat. NH4Cl aq. The whole mixture was extracted with AcOEt, and the AcOEt extract was successively washed with sat. NH4Cl aq., NaHCO3 aq., and brine. Concentration of the AcOEt extract in vacuo gave a crude product which was purified by SiO2 column chromatography (AcOEt : MeOH = 50 : 1) to give a mixture of 5 and 6 (50 mg, 88%, two steps) as a colorless oil.
5: 1H-NMR (600 MHz, CDCl3) δ: 1.16–1.47 (4H, m), 1.58–1.77 (4H, m), 1.86 (3H, s), 1.99–2.14 (4H, m), 2.14–2.23 (5H, m), 2.87–3.10 (6H, m), 3.24–3.40 (2H, m), 3.60 (3H, br s), 4.88 (2H, s), 5.11–5.17 (1H, br s), 5.64 (1H, br s), 5.80 (1H, s), 7.33 (1H, d, J = 2.8 Hz), 7.72 (1H, br s). 13C-NMR (150 MHz, CDCl3) δ: 14.2, 18.8, 20.2, 20.3, 22.8, 24.7, 25.3, 25.6, 25.9, 26.2, 26.3, 26.4, 26. 9, 27.3, 27.5, 27.6, 28.7, 29.1, 29.4, 29.6, 29.8, 29.9, 30.0, 31.6, 32.0, 32.1, 32.7, 32.8, 34.8, 35.8, 35.9, 36.1, 37.0, 46.8, 46.9, 47.4, 48.5, 49.7, 50.1, 54.9, 91.3, 112.4, 112.5, 112.9, 118.0, 118.1, 118.3, 118.4, 120.1, 130.8, 130.9, 132.3, 141.4, 142.0, 142.1, 142.2, 146.3, 146.4, 166.9, 168.3, 168.5, 173.9. IR (KBr) cm−1: 2927, 1644, 1381, 1239, 1072. MS (ESI-TOF) m/z: 518 [M + Na]+. HR-MS (ESI-TOF) m/z: 518.2220 calcd for C25H38N3O3S35ClNa; Found: 518.2212.
6: 1H-NMR (600 MHz, CDCl3) δ: 1.16–1.47 (4H, m), 1.58–1.77 (4H, m), 1.86 (3H, s), 2.00–2.13 (4H, m), 2.13–2.24 (5H, m), 2.89–3.08 (6H, m), 3.24–3.46 (2H, m), 3.61 (3H, s), 4.87 (2H, s), 5.17 (2H, s), 5.63 (1H, s), 5.73–5.85 (1H, m), 7.33 (1H, s), 7.72 (1H, br s). 13C-NMR (150 MHz, CDCl3) δ: 14.4, 18.9, 25.1, 26.1, 27.0, 27.6, 27.7, 28.8, 29.0, 30.1, 33.7, 34.8, 36.1, 37.1, 47.5, 47.7, 48.8, 50.4, 55.0, 91.1, 91.5, 112.4, 113.1, 120.2, 130.9, 132.2, 141.6, 142.3, 166.9, 168.2, 168.5, 174.4. IR (KBr) cm−1: 3019, 1643, 1216, 756. MS (ESI-TOF) m/z: 518 [M + Na]+. HR-MS (ESI-TOF) m/z: 518.2220 calcd for C25H38N3O3S35ClNa; Found: 518.2205.
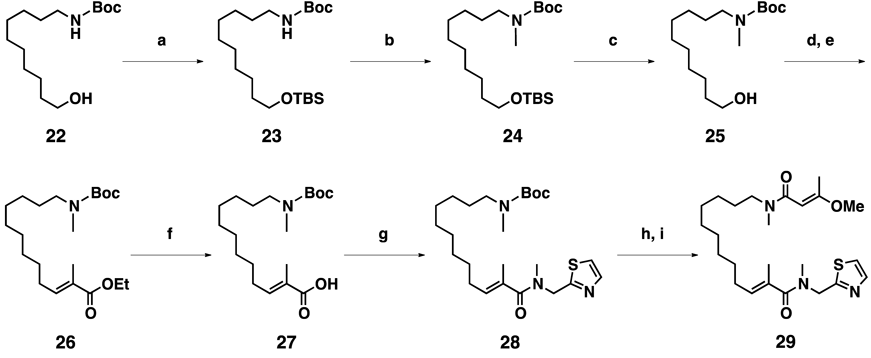
tert-Butyl (10-((tert-Butyldimethylsilyl)oxy)decyl)carbamate (23)Et3N (1.8 mL, 13 mmol, 4.4 equiv.), and TBSCl (759 mg, 5.0 mmol, 1.8 equiv.) were added to a solution of 22 (789 mg, 2.9 mmol) in CH2Cl2 (15 mL), and the mixture was stirred at r.t. for 40 h. Then, the reaction was quenched by the addition of sat. NH4Cl aq. The whole mixture was extracted with AcOEt, and the AcOEt extract was washed with sat. NH4Cl aq. and brine. Concentration of the AcOEt extract in vacuo gave a crude product which was purified by SiO2 column chromatography (hexane : AcOEt = 10 : 1) to give 23 (787 mg, 70%) as a colorless oil.
1H-NMR (600 MHz, CDCl3) δ: –0.02 (3H, s), –0.03 (3H, s), 0.82 (9H, s), 1.20 (12H, br s), 1.29–1.57 (13H, m), 3.02 (2H, br s), 3.52 (2H, td, J = 6.4, 3.4 Hz), 4.70 (1H, br). 13C-NMR (150 MHz, CDCl3) δ: –5.4, –5.2, 18.4, 25.3, 25.8, 25.9, 26.0, 26.2, 26.6, 26.8, 27.4, 27.8, 28.4, 29.1, 29.3, 29.4, 29.5, 29.6, 30.1, 32.9, 40.6, 63.3, 78.8, 156.0. IR (KBr) cm−1: 3356 (br), 2929, 1702, 1514, 1253, 1100, 837. MS (ESI-TOF) m/z: 410 [M + Na]+. HR-MS (ESI-TOF) m/z: 410.3066 calcd for C21H45NO3SiNa; Found: 410.3082.
tert-Butyl (10-((tert-Butyldimethylsilyl)oxy)decyl)(methyl)carbamate (24)NaH (about 60% oil suspension, 80 mg, 2.0 mmol, 1.1 equiv.) was added to a solution of 23 (707 mg, 1.8 mmol) in N,N-dimethylformamide (DMF, 15 mL) at 0°C, and the mixture was stirred at 0°C for 1 h. Iodomethane (250 µL, 4.0 mmol, 2.2 equiv.) was added to the amide solution, and the resulting mixture was stirred at r.t. for 12 h. Then, the reaction was quenched by the addition of sat. NH4Cl aq. The whole mixture was extracted with AcOEt, and the AcOEt extract was washed with NH4Cl aq. and brine. Concentration of the AcOEt extract in vacuo gave a crude product which was purified by SiO2 column chromatography (hexane : AcOEt = 10 : 1) to give 24 (676 mg, 92%) as a colorless oil.
1H-NMR (600 MHz, CDCl3) δ: 0.00 (6H, s), 0.84 (9H, s), 1.23 (12H, br s), 1.38–1.61 (13H, m), 2.78 (3H, s), 3.13 (2H, br s), 3.54 (2H, t, J = 6.6 Hz). 13C-NMR (150 MHz, CDCl3) δ: –5.4, 18.3, 25.7, 25.9, 26.6, 27.5, 27.8, 28.4, 29.3, 29.5, 32.8, 33.9, 48.4, 48.7, 63.2, 78.85, 155.7. IR (KBr) cm−1: 2929, 1686, 1394, 1168, 758. MS (ESI-TOF) m/z: 424 [M + Na]+. HR-MS (ESI-TOF) m/z: 424.3223 calcd for C22H47NO3SiNa; Found: 424.3225.
tert-Butyl (10-Hydroxydecyl)(methyl)carbamate (25)TBAF solution (1 M in THF, 4.4 mL) was added to a solution of 24 (676 mg, 1.7 mmol) in THF (15 mL) at r.t., and the mixture was stirred at r.t. for 8 h. The reaction was quenched by the addition of sat. NH4Cl aq., and the whole mixture was extracted with AcOEt. The AcOEt extract was washed with sat. NH4Cl aq. and brine. Concentration of the AcOEt extract in vacuo gave a crude product, which was purified by SiO2 column chromatography (Hexane : AcOEt = 3 : 1) to give 25 (461 mg, 95%) as a colorless oil.
1H-NMR (600 MHz, CDCl3) δ: 1.02–1.26 (12H, m), 1.29–1.41 (11H, m), 1.44 (2H, dd, J = 13.7, 7.5 Hz), 2.71 (3H, s), 2.84 (1H, br), 3.07 (2H, br s), 3.49 (2H, td, J = 6.7, 3.1 Hz). 13C-NMR (150 MHz, CDCl3) δ: 25.5, 25.7, 26.6, 27.5, 27.8, 28.0, 28.4, 29.3, 29.4, 29.5, 32.7, 33.9, 48.4, 48.7, 62.5, 67.8, 79.0, 155.8. IR (KBr) cm−1: 3448 (br), 2927, 1697, 1397, 1165. MS (ESI-TOF) m/z: 310 [M + Na]+. HR-MS (ESI-TOF) m/z: 310.2358 calcd for C16H33NO3Na; Found: 310.2371.
Ethyl (E)-12-((tert-Butoxycarbonyl)(methyl)amino)-2-methyldodec-2-enoate (26)Iodobenzene diacetate (562 mg, 1.7 mmol, 2.0 equiv.) and TEMPO (14 mg, 0.087 mmol, 0.1 equiv.) were added to a solution of 25 (251 mg, 0.87 mmol) in CH2Cl2 (10 mL), and the mixture was stirred at r.t. for 13 h. Then, toluene (10 mL) and 19 (1.26 g, 3.5 mmol, 4.0 equiv.) were added to the aldehyde solution, and the resulting mixture was stirred at 100°C for 12 h. The reaction was quenched by the addition of Na2S2O3/NaHCO3 aq., and the whole mixture was extracted with AcOEt. The AcOEt extract was successively washed with Na2S2O3/NaHCO3 aq. and brine. Concentration of the AcOEt extract in vacuo gave a crude product which was purified by SiO2 column chromatography (hexane : AcOEt = 10 : 1) to give 26 (291 mg, 90%) as a colorless oil.
1H-NMR (600 MHz, CDCl3) δ: 0.58–1.38 (26H, m), 1.52–2.15 (5H, m), 2.52–2.73 (3H, br s), 2.99 (2H, br s), 3.82–4.20 (2H, m), 6.46–6.64 (1H, br s). 13C-NMR (150 MHz, CDCl3) δ: 12.1, 13.9, 14.0, 14.1, 17.0, 20.4, 26.4, 27.3, 27.6, 28.2, 28.4, 29.1, 29.2, 29.3, 33.7, 48.1, 48.5, 59.9, 60.6, 76.9, 77.2, 77.4, 78.6, 127.4, 133.1, 141.9, 142.8, 155.4, 167.6. IR (KBr) cm−1: 2928, 1699, 1365, 1249, 1166. MS (ESI-TOF) m/z: 392 [M + Na]+. HR-MS (ESI-TOF) m/z: 392.2777 calcd for C21H39NO4Na; Found: 392.2787.
(E)-12-((tert-Butoxycarbonyl)(methyl)amino)-2-methyldodec-2-enoic Acid (27)LiOH (94 mg, 3.9 mmol, 5.0 equiv.) was added to a solution of 26 (291 mg, 0.79 mmol) in THF : MeOH : H2O (1 : 1 : 1, 10 mL), and the mixture was stirred at r.t. for 24 h. The reaction was quenched by the addition of 5% HCl, and the whole mixture was extracted with AcOEt. Concentration of the AcOEt extract in vacuo gave a crude product which was purified by SiO2 column chromatography (hexane : AcOEt = 1 : 1) to give 27 (260 mg, 97%) as a colorless oil.
1H-NMR (600 MHz, CDCl3) δ: 1.22 (10H, br), 1.27–1.82 (13H, m), 2.09 (2H, dd, J = 14.6, 7.3 Hz), 2.74 (2H, s), 3.09 (2H, s), 6.80 (1H, t, J = 7.3 Hz), 11.62 (1H, br). 13C-NMR (150 MHz, CDCl3) δ: 11.7, 20.3, 26.4, 27.3, 27.6, 28.2, 28.3, 28.6, 29.06, 29.11, 29.2, 29.5, 33.8, 48.2, 48.6, 78.9, 126.1, 126.9, 144.6, 145.8, 155.7, 172.9. IR (KBr) cm−1: 2927, 1693, 1394, 1166. MS (ESI-TOF) m/z: 364 [M + Na]+. HR-MS (ESI-TOF) m/z: 364.2464 calcd for C19H35NO4Na; Found: 364.2452.
tert-Butyl (E)-Methyl(11-methyl-12-(methyl(thiazol-2-ylmethyl)amino)-12-oxododec-10-en-1-yl)carbamate (28)HOBt (29 mg, 0.21 mmol, 1.5 equiv.), amine 20 (27 mg, 0.21 mmol, 1.5 equiv.), Et3N (180 µL, 1.3 mmol, 6.0 equiv.), and EDCI·HCl (40 mg, 0.21 mmol, 1.5 equiv.) were added to a solution of 27 (48 mg, 0.14 mmol) in CH2Cl2 (5 mL), and the mixture was stirred at r.t. for 12 h. Then, the reaction was quenched by the addition of sat. NH4Cl aq. The whole mixture was extracted with AcOEt, and the AcOEt extract was successively washed with sat. NH4Cl aq., NaHCO3 aq., and brine. Concentration of the AcOEt extract in vacuo gave a crude product which was purified by SiO2 column chromatography (hexane : AcOEt = 1 : 1) to give 28 (56 mg, 88%) as a colorless oil.
1H-NMR (600 MHz, CDCl3) δ: 1.16 (10H, m), 1.28 (2H, s), 1.36 (11H, m), 1.76 (3H, s), 2.00 (2H, br s), 2.72 (3H, s), 2.94 (3H, s), 3.08 (2H, s), 4.78 (2H, s), 5.55 (1H, br s), 7.24 (1H, br s), 7.62 (1h, br s). 13C-NMR (150 MHz, CDCl3) δ: 14.1, 20.0, 20.8, 26.6, 27.5, 27.8, 28.2, 28.4, 28.5, 28.7, 29.0, 29.1, 29.2, 29.3, 29.4, 29.6, 29.7, 32.3, 33.9, 35.5, 36.9, 47.5, 48.3, 48.7, 51.8, 52.8, 78.9, 119.7, 119.9, 120.1, 129.9, 130.5, 130.8, 132.4, 142.0, 142.8, 155.7, 166.2, 166.8, 172.1, 173.7. IR (KBr) cm−1: 2929, 1683, 1398, 1216, 755. MS (ESI-TOF) m/z: 474 [M + Na]+. HR-MS (ESI-TOF) m/z: 474.2766 calcd for C24H41N3O3SNa; Found: 474.2763.
(E)-12-((E)-3-Methoxy-N-methylbut-2-enamido)-N,2-dimethyl-N-(thiazol-2-ylmethyl)dodec-2-enamide (29)TFA (165 µL, 2.2 mmol, 50 equiv.) was added to a solution of 28 (20 mg, 0.044 mmol) in CH2Cl2 (2 mL), and the mixture was stirred at r.t. for 1 h. Then, the reaction was quenched by the addition of sat. NaHCO3 aq. The whole mixture was extracted with AcOEt, and the AcOEt extract was washed with sat. NaHCO3 aq. and brine. Concentration of the AcOEt extract in vacuo gave an amine, which was used in the next reaction without further purification. The above crude product was dissolved to CH2Cl2 (1 mL). HOBt (10 mg, 0.062 mmol, 1.4 equiv.), 21 (23 mg, 0.18 mmol, 4.0 equiv.), Et3N (52 µL, 0.37 mmol, 6.0 equiv.), and EDCI·HCl (12 mg, 0.062 mmol, 1.4 equiv.) were added to the solution, and the resulting mixture was stirred for 12 h at r.t. Then, the reaction was quenched by the addition of sat. NH4Cl aq. The whole mixture was extracted with AcOEt, and the AcOEt extract was successively washed with sat. NH4Cl aq., NaHCO3 aq., and brine. Concentration of the AcOEt extract in vacuo gave a crude product which was purified by SiO2 column chromatography (hexane : AcOEt = 1 : 3) to give 29 (14 mg, 69%, two steps) as a colorless oil.
1H-NMR (600 MHz, CDCl3) δ: 1.08–1.42 (12H, m), 1.44–1.57 (2H, m), 1.83 (3H, s), 2.01–2.10 (2H, m), 2.11–2.23 (3H, m), 2.81–3.08 (6H, m), 3.27 (1H, br s), 3.35 (1H, br s), 3.57 (3H, s), 4.85 (2H, s), 5.14 (1H, d, J = 9.9 Hz), 5.62 (1H, br s), 7.30 (1H, br s), 7.69 (1H, br s). 13C-NMR (150 MHz, CDCl3) δ: 14.8, 18.8, 23.1, 23.8, 26.8, 27.0, 27.6, 27.7, 28.5, 28.9, 29.4, 29.5, 29.6, 30.4, 31.0, 33.6, 35.7, 36.0, 38.8, 47.7, 50.6, 54.85, 54.90, 68.2, 91.4, 91.5, 120.1, 128.9, 130.6, 130.7, 131.0, 132.5, 132.6, 142.2, 166.4, 166.9, 167.8, 168.0, 168.1, 168.2, 172.3, 173.8. IR (KBr) cm−1: 2926, 1648, 1381, 1238, 1140, 1073. MS (ESI-TOF) m/z: 472 [M + Na]+. HR-MS (ESI-TOF) m/z: 472.2610 calcd for C24H39N3O3SNa; Found: 472.2600.
tert-Butyl ((10E)-4-(Hydroxyimino)-11-methyl-12-(methyl(thiazol-2-ylmethyl)amino)-12-oxododec-10-en-1-yl)(methyl)carbamate (30)Hydroxylamine hydrochloride (2.7 mg, 0.026 mmol, 1.5 equiv.) and AcONa (3.2 mg, 0.039 mmol) were added to a solution of 16 in MeOH (0.5 mL), and the mixture was stirred at r.t. for 1 h. Concentration in vacuo and purification of the residue by SiO2 column chromatography (hexane : AcOEt = 1 : 3) gave 30 (10 mg, 83%) as a colorless oil.
1H-NMR (600 MHz, CDCl3) δ: 1.18–1.51 (15H, m), 1.64–1.77 (2H, m), 1.83 (3H, s), 2.03–2.22 (4H, m), 2.22–2.41 (2H, m), 2.82 (3H, br s), 3.02 (3H, br s), 3.20 (2H, br s), 4.87 (3H, d, J = 4.8 Hz), 5.62 (1H, d, J = 6.2 Hz), 7.31 (1H, br s), 7.71 (1H, br s), 8.78 (1H, br). 13C-NMR (150 MHz, CDCl3) δ: 14.2, 20.0, 20.1, 22.8, 23.7, 24.1, 24.3, 24.6, 25.0, 25.6, 26.0, 27.51, 27.54, 27.6, 28.56, 28.58, 28.6, 28.8, 29.4, 29.6, 29.8, 31.7, 34.0, 34.3, 37.1, 48.2, 48.5, 48.7, 49.1, 53.0, 79.4, 120.2, 130.9, 132.3, 142.2, 155.9, 160.7, 167.0, 174.1. IR (KBr) cm−1: 3332 (br), 2929, 1692, 1396, 1169, 755. MS (ESI-TOF) m/z: 503 [M + Na]+. HR-MS (ESI-TOF) m/z: 503.2668 calcd for C24H40N4O4SNa; Found: 503.2669.
(2E)-9-(Hydroxyimino)-12-((E)-3-methoxy-N-methylbut-2-enamido)-N,2-dimethyl-N-(thiazol-2-ylmethyl)dodec-2-enamide (31)TFA (49 µL, 0.43 mmol, 20 equiv.) was added to a solution of 30 (10 mg, 0.022 mmol) in CH2Cl2 (1 mL), and the mixture was stirred at r.t. for 3 h. Then, the reaction was quenched by the addition of sat. NaHCO3 aq. The whole mixture was extracted with AcOEt, and the AcOEt extract was washed with sat. NaHCO3 aq. and brine. Concentration of the AcOEt extract in vacuo gave an amine, which was used in the next reaction without further purification. The above crude product was dissolved to CH2Cl2 (1 mL). HOBt (4.3 mg, 0.029 mmol, 1.3 equiv.), 21 (2.2 mg, 0.019 mmol, 0.9 equiv.), Et3N (12 µL, 0.85 mmol, 3.9 equiv.), and EDCI·HCl (6.1 mg, 0.029 mmol, 1.3 equiv.) were added to the solution, and the resulting mixture was stirred at r.t. for 9 h. Then, the reaction was quenched by the addition of sat. NH4Cl aq. The whole mixture was extracted with AcOEt, and the AcOEt extract was successively washed with sat. NH4Cl aq., NaHCO3 aq., and brine. Concentration of the AcOEt extract in vacuo gave a crude product which was purified by SiO2 column chromatography (AcOEt : MeOH = 30 : 1) to give 31 (3.5 mg, 34%, two steps) as a colorless oil.
1H-NMR (600 MHz, CDCl3) δ: 1.12–1.55 (6H, m), 1.69–1.93 (5H, m), 1.96–2.46 (9H, m), 2.86–3.10 (6H, m), 3.30–3.47 (2H, m), 3.53–3.64 (3H, m), 4.83–4.94 (2H, m), 5.16 (1H, br s), 5.63 (1H, br s), 7.32 (1H, br s), 7.72 (1H, br s), 8.42 (1H, br s). 13C-NMR (150 MHz, CDCl3) δ: 14.3, 18.9, 23.6, 24.3, 24.6, 25.0, 25.2, 25.6, 25.9, 27.5, 27.6, 28.6, 29.2, 29.3, 29.8, 30.9, 31.6, 33.6, 34.0, 36.06, 36.14, 37.1, 47.4, 47.7, 48.5, 49.9, 50.6, 55.0, 91.2, 91.4, 120.2, 130.9, 132.4, 142.2, 142.4, 160.3, 161.1, 168.2, 168.6, 174.1. IR (KBr) cm−1: 3306 (br), 2925, 1641, 1384, 1240. MS (ESI-TOF) m/z: 501 [M + Na]+. HR-MS (ESI-TOF) m/z: 501.2511 calcd for C24H38N4O4SNa; Found: 501.2503.
tert-Butyl (E)-Methyl(3-(2-(7-methyl-8-oxo-8-((thiazol-2-ylmethyl)amino)oct-6-en-1-yl)-1,3-dithian-2-yl)propyl)carbamate (32)HOBt (38 mg, 0.28 mmol, 1.4 equiv.), 2-(aminomethyl)thiazole (37, 35 mg, 0.30 mmol, 1.5 equiv.), Et3N (160 µL, 1.1 mmol, 4.0 equiv.), and EDCI·HCl (54 mg, 0.28 mmol, 1.4 equiv.) were added to a solution of 14 (90 mg, 0.20 mmol) in CH2Cl2 (5 mL), and the mixture was stirred at r.t. for 10 h. Then, the reaction was quenched by the addition of sat. NH4Cl aq. The whole mixture was extracted with AcOEt, and the AcOEt extract was successively washed with sat. NH4Cl aq., NaHCO3 aq., and brine. Concentration of the AcOEt extract in vacuo gave a crude product which was purified by SiO2 column chromatography (hexane : AcOEt = 1 : 3) to give 32 (87 mg, 80%) as a colorless oil.
1H-NMR (500 MHz, CDCl3) δ: 1.24 (2H, d, J = 7.1 Hz), 1.31–1.38 (13H, m), 1.49–1.76 (7H, m), 1.78 (3H, s), 1.84 (2H, d, J = 4.6 Hz), 2.06 (2H, dd, J = 14.5, 7.2 Hz), 2.69 (4H, d, J = 4.7 Hz), 2.75 (3H, s), 3.13 (2H, br s), 4.70 (2H, d, J = 5.7 Hz), 6.35 (1H, t, J = 7.1 Hz), 7.00 (1H, br s), 7.20 (1H, d, J = 3.2 Hz), 7.60 (1H, d, J = 3.1 Hz). 13C-NMR (150 MHz, CDCl3) δ: 12.6, 22.4, 22.7, 23.8, 25.3, 25.7, 25.9, 26.0, 27.7, 28.0, 28.2, 28.4, 29.1, 29.3, 34.0, 34.1, 35.0, 38.2, 41.0, 47.9, 48.7, 52.8, 60.3, 79.1, 119.5, 130.2, 137.0, 142.0, 142.1, 155.6, 167.9, 169.2. IR (KBr) cm−1: 3330 (br), 2933, 1692, 1523, 1394, 1150. MS (ESI-TOF) m/z: 564 [M + Na]+. HR-MS (ESI-TOF) m/z: 564.2364 calcd for C26H43N3O3S3Na; Found: 564.2391.
tert-Butyl (E)-Methyl(11-methyl-4,12-dioxo-12-((thiazol-2-ylmethyl)amino)dodec-10-en-1-yl)carbamate (33)Iodine (77 mg, 0.30 mmol, 2.0 equiv.) was added to a solution of 32 (82 mg, 0.15 mmol) in a mixture of CH3CN (6 mL) and sat. NaHCO3 aq. (3 mL) at 0°C, and the mixture was stirred at 0°C for 3 h. The reaction was quenched by the addition of Na2S2O3/NaHCO3 aq., and the whole mixture was extracted with AcOEt. Then, the AcOEt extract was washed with Na2S2O3/NaHCO3 aq. and brine. Concentration of the AcOEt extract in vacuo gave a crude product which was purified by SiO2 column chromatography (hexane : AcOEt = 1 : 4) to give 33 (27 mg, 40%) as a colorless oil.
1H-NMR (600 MHz, CDCl3) δ: 1.23 (2H, br), 1.37 (11H, m), 1.49 (2H, br s), 1.71 (2H, br), 1.80 (3H, s), 2.08 (2H, br s), 2.20–2.45 (4H, br), 2.74 (3H, s), 3.13 (2H, br s), 4.74 (2H, br), 6.36 (1H, br s), 6.97–7.02 (1H, br), 7.24 (1H, br s), 7.63 (1H, br s). 13C-NMR (150 MHz, CDCl3) δ: 12.7, 21.5, 21.8, 23.5, 28.1, 28.4, 28.5, 28.8, 29.9, 34.0, 38.3, 39.5, 41.0, 42.6, 47.5, 48.1, 63.4, 79.2, 119.7, 130.3, 137.1, 141.9, 155.9, 168.1, 169.3, 210.1, 210.4. IR (KBr) cm−1: 3341 (br), 2931, 1692, 1523, 1395, 1168. MS (ESI-TOF) m/z: 474 [M + Na]+. HR-MS (ESI-TOF) m/z: 474.2402 calcd for C23H37N3O4SNa; Found: 474.2403.
tert-Butyl ((10E)-4-(Chloromethylene)-11-methyl-12-oxo-12-((thiazol-2-ylmethyl)amino)dodec-10-en-1-yl)(methyl)carbamate (34)LHMDS solution (1.0 M in THF, 170 µL, 0.17 mmol, 2.8 equiv.) was added to a solution of (chloromethyl)triphenylphosphonium chloride (63 mg, 0.18 mmol, 3.0 equiv.) in anhydrous THF (3 mL), and the mixture was stirred at r.t. for 1 h. A solution of 33 (27 mg, 0.060 mmol) in anhydrous THF (0.5 mL) was added dropwise to the ylide solution via cannula at r.t., and the resulting mixture was stirred at r.t. for 4 h. Then, the reaction was quenched by the addition of sat. NH4Cl aq. The whole mixture was extracted with AcOEt, and the AcOEt extract was washed with NH4Cl aq. and brine. Concentration of the AcOEt extract in vacuo gave a crude product which was purified by SiO2 column chromatography (hexane : AcOEt = 1 : 3) to give 34 (23 mg, 79%) as a colorless oil.
1H-NMR (600 MHz, CDCl3) δ: 1.20–1.44 (15H, m), 1.58 (2H, br), 1.84 (3H, s), 1.96–2.04 (2H, m), 2.08–2.19 (4H, m), 2.80 (3H, m), 3.15 (2H, br s), 4.77 (2H, d, J = 5.5 Hz), 5.76 (1H, br s), 6.37–6.44 (1H, br), 6.81 (1H, br s), 7.26 (1H, d, J = 2.4 Hz), 7.67 (1H, br s). 13C-NMR (150 MHz, CDCl3) δ: 12.7, 25.1, 25.5, 25.9, 26.9, 27.4, 27.5, 28.3, 28.4, 28.52, 28.54, 29.0, 29.2, 30.0, 31.9, 34.3, 34.8, 41.2, 48.2, 48.5, 48.9, 79.3, 79.4, 112.4, 119.7, 128.5, 128.6, 130.2, 130.3, 132.0, 132.07, 132.14, 137.3, 137.4, 141.9, 142.2, 155.8, 155.8, 167.8, 169.2, 169.3. IR (KBr) cm−1: 3331 (br), 2930, 1693, 1523, 1394, 1166. MS (ESI-TOF) m/z: 506 [M + Na]+. HR-MS (ESI-TOF) m/z: 506.2220 calcd for C24H38N3O3S35ClNa; Found: 506.2195.
tert-Butyl ((10E)-4-(Chloromethylene)-11-methyl-12-oxo-12-((thiazol-2-ylmethyl)amino)dodec-10-en-1-yl)(methyl)carbamate (35)TFA (65 µL, 0.87 mmol, 20 equiv.) was added to a solution of 34 (21 mg, 0.044 mmol) in CH2Cl2 (1 mL), and the mixture was stirred at r.t. for 1 h. Then, the reaction was quenched by the addition of sat. NaHCO3 aq. The whole mixture was extracted with AcOEt, and the AcOEt extract was washed with sat. NaHCO3 aq. and brine. Concentration of the AcOEt extract in vacuo gave an amine, which was used in the next reaction without further purification. The above crude product was dissolved to CH2Cl2 (1 mL). HOBt (11 mg, 0.078 mmol, 1.8 equiv.), 21 (9.0 mg, 0.078 mmol, 1.8 equiv.), Et3N (29 µL, 0.21 mmol, 4.8 equiv.), and EDCI·HCl (15 mg, 0.078 mmol, 1.8 equiv.) were added to the solution, and the resulting mixture was stirred at r.t. for 24 h. Then, the reaction was quenched by the addition of sat. NH4Cl aq. The whole mixture was extracted with AcOEt, and the AcOEt extract was washed with sat. NH4Cl aq., NaHCO3 aq., and brine. Concentration of the AcOEt extract in vacuo gave a crude product which was purified by SiO2 column chromatography (hexane : AcOEt = 1 : 5) to give 35 and 36 (23 mg, 97%, two steps) as a colorless oil.
35: 1H-NMR (500 MHz, CDCl3) δ: 1.02–1.70 (8H, br), 1.87 (3H, s), 2.06 (3H, dd, J = 10.0, 5.1 Hz), 2.10–2.27 (6H, br), 2.97 (3H, br), 3.20–3.44 (2H, br), 3.60 (3H, s), 4.82 (2H, d, J = 5.7 Hz), 5.14 (1H, s), 5.81 (1H, s), 6.44 (1H, t, J = 7.4 Hz), 6.69 (1H, br s), 7.29 (1H, d, J = 3.3 Hz), 7.71 (1H, d, J = 3.3 Hz). 13C-NMR (150 MHz, CDCl3) δ: 12.8, 18.9, 27.0, 28.4, 28.6, 29.2, 30.0, 32.3, 36.2, 41.3, 47.5, 49.9, 55.0, 77.4, 91.3, 112.5, 119.6, 130.3, 137.5, 142.4, 165.6, 168.7, 169.4. IR (KBr) cm−1: 3326 (br), 2931, 1635, 1522, 1360, 1139. In Mass mesurement, the spectrum of 3-oxobutanoic amide derivative which decomposed from compound 35 was obtained. MS (ESI-TOF) m/z: 490 [M + Na]+. HR-MS (ESI-TOF) m/z: 490.1910 calcd for C23H34N3O3S35ClNa; Found: 490.1910.
Ethyl (E)-12-((tert-Butoxycarbonyl)(methyl)amino)-2-methyl-9-oxododec-2-enoate (38)Iodine (49 mg, 0.19 mmol, 3.4 equiv.) was added to a solution of 15 (27 mg, 0.057 mmol) in a mixture of CH3CN (2 mL) and sat. NaHCO3 aq. (1 mL) at 0°C, and the mixture was stirred at 0°C for 1 h. The reaction was quenched by the addition of Na2S2O3/NaHCO3 aq., and the whole mixture was extracted with AcOEt. Then, the AcOEt extract was washed with Na2S2O3/NaHCO3 aq. and brine. Concentration of the AcOEt extract in vacuo gave a crude product which was purified by SiO2 column chromatography (hexane : AcOEt = 3 : 1) to give 38 (14 mg, 62%) as a colorless oil.
1H-NMR (600 MHz, CDCl3) δ: 1.21–1.32 (5H, m), 1.43 (11H, s), 1.52–1.60 (2H, m), 1.67–1.82 (5H, m), 2.10–2.17 (2H, m), 2.34–2.42 (4H, m), 2.80 (3H, s), 3.19 (2H, s), 4.16 (2H, q, J = 7.1 Hz), 6.71 (1H, t, J = 7.3 Hz). 13C-NMR (150 MHz, CDCl3) δ: 12.5, 14.4, 14.5, 21.6, 21.9, 23.6, 26.1, 28.5, 28.6, 28.7, 29.0, 29.5, 34.1, 39.6, 42.8, 47.7, 48.2, 60.1, 60.5, 79.4, 128.0, 142.1, 142.8, 155.9, 168.4, 210.2, 210.6. IR (KBr) cm−1: 2932, 1697, 1365, 1171. MS (ESI-TOF) m/z: 406 [M + Na]+. HR-MS (ESI-TOF) m/z: 406.2569 calcd for C21H37NO5Na; Found: 406.2566.
Ethyl (2E)-12-((tert-Butoxycarbonyl)(methyl)amino)-9-(chloromethylene)-2-methyldodec-2-enoate (39)LHMDS solution (1.0 M in THF, 170 µL, 0.17 mmol, 4.8 equiv.) was added to a solution of (chloromethyl)triphenylphosphonium chloride (61 mg, 0.18 mmol, 5.0 equiv.) in anhydrous THF (2 mL), and the mixture was stirred for 1 h at r.t. A solution of 38 (14 mg, 0.035 mmol) in anhydrous THF (0.5 mL) was added dropwise to the ylide solution via cannula at r.t., and the resulting mixture was stirred at r.t. for 1 h. Then, the reaction was quenched by the addition of sat. NH4Cl aq., and the whole mixture was extracted with AcOEt, and the AcOEt extract was washed with NH4Cl aq. and brine. Concentration of the AcOEt extract in vacuo gave a crude product which was purified by SiO2 column chromatography (hexane : AcOEt = 5 : 1) to give 39 (11 mg, 75%) as a colorless oil.
1H-NMR (600 MHz, CDCl3) δ: 1.29 (3H, t, J = 7.1 Hz), 1.39–1.51 (11H, m), 1.55–1.69 (6H, m), 1.82 (3H, s), 2.03 (2H, t, J = 7.0 Hz), 2.12–2.25 (4H, m), 2.83 (3H, s), 3.18 (2H, br s), 4.19 (2H, q, J = 7.1 Hz), 5.81 (s, 1H), 6.72–6.78 (1H, m). 13C-NMR (150 MHz, CDCl3) δ: 12.5, 14.5, 19.2, 20.8, 26.7, 27.0, 27.6, 28.5, 28.6, 28.7, 29.1, 29.3, 29.9, 30.1, 32.0, 34.3, 34.6, 48.4, 48.6, 49.0, 60.6, 79.4, 112.5, 127.9, 128.0, 129.8, 134.9, 135.3, 142.2, 155.9, 168.4. IR (KBr) cm−1: 2930, 1672, 1428, 1113, 704. MS (ESI-TOF) m/z: 438 [M + Na]+. HR-MS (ESI-TOF) m/z: 438.2387 calcd for C22H38NO435ClNa; Found: 438.2389.
Ethyl (2E,9E)-9-(Chloromethylene)-12-((E)-3-methoxy-N-methylbut-2-enamido)-2-methyldodec-2-enoate (40)TFA (80 µL, 1.1 mmol, 47 equiv.) was added to a solution of 39 (11 mg, 0.026 mmol) in CH2Cl2 (1 mL), and the mixture was stirred at r.t. for 1.5 h. Then, the reaction was quenched by the addition of sat. NaHCO3 aq. The whole mixture was extracted with AcOEt, and the AcOEt extract was washed with sat. NaHCO3 aq. and brine. Concentration of the AcOEt extract in vacuo gave an amine, which was used in the next reaction without further purification. The above crude product was dissolved to CH2Cl2 (1 mL). HOBt (5.0 mg, 0.037 mmol, 1.6 equiv.), 21 (4.1 mg, 0.035 mmol, 1.6 equiv.), Et3N (31 µL, 0.22 mmol, 9.8 equiv.), and EDCI·HCl (7.1 mg, 0.037 mmol, 1.6 equiv.) were added to the solution, and the resulting mixture was stirred at r.t. for 7 h. Then, the reaction was quenched by the addition of sat. NH4Cl aq. The whole mixture was extracted with AcOEt, and the AcOEt extract was washed with sat. NH4Cl aq., NaHCO3 aq., and brine. Concentration of the AcOEt extract in vacuo gave a crude product which was purified by SiO2 column chromatography (hexane : AcOEt = 3 : 1) to give 40 and 41 (6.8 mg, 72%, two steps) as a colorless oil.
40: 1H-NMR (600 MHz, CDCl3) δ: 1.09–1.78 (12H, m), 1.82 (3H, s), 2.06 (2H, m), 2.11–2.28 (6H, m), 2.94–3.00 (3H, m), 3.29–3.38 (2H, br), 3.60 (3H, s), 4.18 (2H, q, J = 7.1 Hz), 5.15 (1H, s), 5.81 (1H, s), 6.74 (1H, td, J = 7.5, 1.3 Hz). 13C-NMR (150 MHz, CDCl3) δ: 12.5, 14.4, 18.9, 25.7, 26.2, 27.0, 28.5, 28.7, 29.3, 30.1, 47.5, 55.0, 60.6, 76.8, 77.4, 77.5, 91.3, 112.5, 142.2, 142.4, 168.5, 168.6. IR (KBr) cm−1: 2929, 1651, 1442, 1241, 1119. MS (ESI-TOF) m/z: 436 [M + Na]+. HR-MS (ESI-TOF) m/z: 436.2231 calcd for C22H36NO435ClNa; Found: 436.2214.
tert-Butyl (E)-Methyl(3-(2-(7-methyl-8-(methyl(naphthalen-1-ylmethyl)amino)-8-oxooct-6-en-1-yl)-1,3-dithian-2-yl)propyl)carbamate (42)HOBt (43 mg, 0.32 mmol, 1.4 equiv.), N-methyl-1-naphthylmethylamine (47, 78 mg, 0.45 mmol, 2.0 equiv.), Et3N (265 µL, 1.9 mmol, 8.4 equiv.), and EDCI·HCl (61 mg, 0.32 mmol, 1.4 equiv.) were added to a solution of 14 (101 mg, 0.23 mmol) in CH2Cl2 (5 mL), and the mixture was stirred at r.t. for 8 h. Then, the reaction was quenched by the addition of sat. NH4Cl aq. The whole mixture was extracted with AcOEt, and the AcOEt extract was successively washed with sat. NH4Cl aq., NaHCO3 aq., and brine. Concentration of the AcOEt extract in vacuo gave a crude product which was purified by SiO2 column chromatography (hexane : AcOEt = 3 : 1) to give 42 (124 mg, 91%) as a colorless oil.
1H-NMR (600 MHz, CDCl3) δ: 0.73–1.48 (15H, m), 1.51–1.62 (2H, br s), 1.65–1.92 (9H, m), 1.93–2.08 (2H, br s), 2.60–2.74 (10H, br), 3.16 (2H, br s), 5.02 (2H, s), 5.51 (1H, br s), 7.24–7.35 (1H, br s), 7.35–7.55 (3H, m), 7.72–8.01 (3H, m). 13C-NMR (150 MHz, CDCl3) δ: 14.2, 22.7, 23.7, 25.2, 25.3, 25.4, 25.8, 27.3, 28.4, 28.5, 29.3, 34.0, 34.8, 35.8, 38.2, 48.7, 52.8, 79.1, 104.9, 123.8, 125.1, 125.8, 125.9, 126.3, 126.4, 128.5, 131.1, 132.4, 133.7, 155.6, 173.3. IR (KBr) cm−1: 2936, 1685, 1616, 1398, 1216, 1153, 755. MS (ESI-TOF) m/z: 621 [M + Na]+. HR-MS (ESI-TOF) m/z: 621.3161 calcd for C34H50N2O3S2Na; Found: 621.3172.
tert-Butyl (E)-Methyl(11-methyl-12-(methyl(naphthalen-1-ylmethyl)amino)-4,12-dioxododec-10-en-1-yl)carbamate (43)Iodine (105 mg, 0.41 mmol, 2.0 equiv.) was added to a solution of 42 (124 mg, 0.21 mmol) in a mixture of CH3CN (6 mL) and sat. NaHCO3 aq. (3 mL) at 0°C, and the mixture was stirred at 0°C for 1 h. The reaction was quenched by the addition of Na2S2O3/NaHCO3 aq., and the whole mixture was extracted with AcOEt. Then, the AcOEt extract was washed with Na2S2O3/NaHCO3 aq. and brine. Concentration of the AcOEt extract in vacuo gave a crude product which was purified by SiO2 column chromatography (hexane : AcOEt = 1 : 1) to give 43 (60 mg, 57%) as a colorless oil.
1H-NMR (600 MHz, CDCl3) δ: 0.61–1.63 (15H, m), 1.63–1.78 (2H, m), 1.83 (3H, s), 1.97 (2H, m), 2.31 (4H, br s), 2.56–3.09 (6H, m), 3.16 (2H, br s), 5.03 (2H, s), 5.51 (1H, br s), 7.32 (1H, br s), 7.41 (1H, br s), 7.44–7.56 (2H, m), 7.77 (1H, d, J = 8.2 Hz), 7.84 (1H, d, J = 7.2 Hz), 8.03 (1H, br s). 13C-NMR (150 MHz, CDCl3) δ: 14.3, 21.7, 23.4, 27.3, 28.4, 28.6, 28.8, 29.7, 34.0, 35.9, 39.4, 42.6, 47.76, 48.00, 52.6, 79.3, 122.3, 122.8, 123.9, 125.3, 126.0, 126.4, 127.1, 128.3, 128.7, 131.1, 131.5, 132.5, 133.8, 155.8, 173.4, 210.1. IR (KBr) cm−1: 2930, 1692, 1622, 1396, 1168. MS (ESI-TOF) m/z: 531 [M + Na]+. HR-MS (ESI-TOF) m/z: 531.3199 calcd for C31H44N2O4Na; Found: 531.3174.
tert-Butyl ((10E)-4-(Chloromethylene)-11-methyl-12-(methyl(naphthalen-1-ylmethyl)amino)-12-oxododec-10-en-1-yl)(methyl)carbamate (44)LHMDS solution (1.0 M in THF, 230 µL, 0.23 mmol, 3.0 equiv.) was added to a solution of (chloromethyl)triphenylphosphonium chloride (80 mg, 0.23 mmol, 3.0 equiv.) in anhydrous THF (2 mL), and the mixture was stirred at r.t. for 0.5 h. A solution of 43 (39 mg, 0.077 mmol) in anhydrous THF (1 mL) was added dropwise to the ylide solution via cannula at r.t. over 5 min, and the resulting mixture was stirred at r.t. for 2 h. Then, the reaction was quenched by the addition of sat. NH4Cl aq. The whole mixture was extracted with AcOEt, and the AcOEt extract was washed with NH4Cl aq. and brine. Concentration of the AcOEt extract in vacuo gave a crude product which was purified by SiO2 column chromatography (hexane : AcOEt = 1 : 1) to give 44 (29 mg, 70%) as a colorless oil.
1H-NMR (600 MHz, CDCl3) δ: 0.74–1.50 (15H, m), 1.56 (2H, br s), 1.71–2.24 (9H, m), 2.60–3.26 (8H, m), 5.07 (2H, s), 5.42–5.83 (2H, m), 7.29–7.54 (4H, m), 7.75–7.90 (2H, m), 8.06 (1H, br s). 13C-NMR (150 MHz, CDCl3) δ: 14.4, 25.3, 25.8, 26.8, 27.4, 27.5, 28.4, 28.5, 28.6, 28.7, 29.1, 30.0, 31.9, 34.3, 34.7, 36.0, 48.4, 52.7, 79.3, 79.4, 112.4, 124.0, 125.3, 126.1, 126.5, 127.3, 128.4, 128.8, 131.3, 132.6, 133.9, 142.0, 155.80, 155.82, 173.5. IR (KBr) cm−1: 2931, 1686, 1616, 1397, 1216, 754. MS (ESI-TOF) m/z: 563 [M + Na]+. HR-MS (ESI-TOF) m/z: 563.3016 calcd for C32H45N2O335ClNa; Found: 563.3020.
(2E,9E)-9-(Chloromethylene)-12-((E)-3-methoxy-N-methylbut-2-enamido)-N,2-dimethyl-N-(naphthalen-1-ylmethyl)dodec-2-enamide (45)TFA (83 µL, 1.1 mmol, 20 equiv.) was added to a solution of 44 (29 mg, 0.057 mmol) in CH2Cl2 (1 mL), and the mixture was stirred at r.t. for 1 h. Then, the reaction was quenched by the addition of sat. NaHCO3 aq. The whole mixture was extracted with AcOEt, and the AcOEt extract was washed with sat. NaHCO3 aq. and brine. Concentration of the AcOEt extract in vacuo gave an amine, which was used in the next reaction without further purification. The above crude product was dissolved to CH2Cl2 (1 mL). HOBt (11 mg, 0.080 mmol, 1.4 equiv.), 21 (9.3 mg, 0.080 mmol, 1.4 equiv.), Et3N (32 µL, 0.23 mmol, 4.0 equiv.), and EDCI·HCl (15 mg, 0.080 mmol, 1.4 equiv.) were added to the solution, and the resulting mixture was stirred at r.t. for 2.5 h. Then, the reaction was quenched by the addition of sat. NH4Cl aq. The whole mixture was extracted with AcOEt, and the AcOEt extract was successively washed with sat. NH4Cl aq., NaHCO3 aq., and brine. Concentration of the AcOEt extract in vacuo gave a crude product which was purified by SiO2 column chromatography (hexane : AcOEt = 1 : 2) to give 45 and 46 (8.0 mg, 26%, two steps) as a colorless oil.
45: 1H-NMR (500 MHz, CDCl3) δ: 1.25–1.66 (8H, br), 1.86 (3H, s), 2.03 (6H, br), 2.18 (3H, s), 2.58–3.12 (6H, br), 3.32 (2H, br), 3.61 (3H, s), 5.07 (2H, s), 5.15 (1H, d, J = 5.9 Hz), 5.57 (1H, s), 5.77 (1H, s), 7.32–7.96 (9H, m). 13C-NMR (150 MHz, CDCl3) δ: 11.1, 14.2, 14.4, 18.9, 23.1, 23.7, 23.8, 25.7, 26.2, 26.9, 27.6, 28.7, 29.0, 29.8, 30.0, 30.5, 31.6, 32.2, 33.5, 36.1, 38.8, 47.4, 49.8, 55.0, 68.3, 91.0, 91.3, 112.4, 113.0, 124.0, 125.4, 126.1, 126.6, 127.2, 128.5, 128.9, 131.0, 131.4, 131.9, 132.5, 133.9, 164.0, 167.9, 174.1. IR (KBr) cm−1: 2930, 1728, 1275, 1123. MS (ESI-TOF) m/z: 561 [M + Na]+. HR-MS (ESI-TOF) m/z: 561.2860 calcd for C32H43N2O335ClNa; Found: 561.2850.
(2E,9E)-9-(Chloromethylene)-N,2-dimethyl-12-(N-methylacetamido)-N-(thiazol-2-ylmethyl)dodec-2-enamide (48)TFA (46 µL, 0.62 mmol, 30 equiv.) was added to a solution of 17 (10 mg, 0.020 mmol) in CH2Cl2 (1 mL), and the mixture was stirred at r.t. for 1 h. Then, the reaction was quenched by the addition of sat. NaHCO3 aq. The whole mixture was extracted with AcOEt, and the AcOEt extract was washed with sat. NaHCO3 aq. and brine. Concentration of the AcOEt extract in vacuo gave an amine, which was used in the next reaction without further purification. The above crude product was dissolved to CH2Cl2 (1 mL). 4-Dimethylaminopyridine (DMAP, 3.8 mg, 0.031 mmol, 1.5 equiv.), Et3N (60 µL, 0.43 mmol, 21 equiv.), and acetyl chloride (10 µL, 0.14 mmol, 6.9 equiv.) were added to the solution, and the resulting mixture was stirred for 1 h at r.t. Then, the reaction was quenched by the addition of sat. NaHCO3 aq. The whole mixture was extracted with AcOEt, and the AcOEt extract was washed with sat. NaHCO3 aq., and brine. Concentration of the AcOEt extract in vacuo gave a crude product which was purified by SiO2 column chromatography (hexane : AcOEt = 1 : 5) to give 48 and 49 (9.1 mg, quant., two steps) as a colorless oil.
48: 1H-NMR (600 MHz, CDCl3) δ: 1.11–1.46 (6H, m), 1.53–1.72 (2H, m), 1.85 (3H, s), 1.97–2.24 (9H, m), 2.86–2.92 (3H, m), 2.93–3.09 (3H, m), 3.18–3.38 (2H, m), 4.86 (2H, s), 5.63 (1H, br s), 5.74–5.83 (1H, m), 7.32 (1H, br s), 7.71 (1H, br s). 13C-NMR (150 MHz, CDCl3) δ: 14.4, 21.4, 22.1, 24.8, 25.4, 25.9, 26.3, 26.9, 27.0, 27.4, 27.48, 27.53, 27.65, 27.67, 28.70, 28.72, 29.0, 29.2, 29.8, 29.97, 30.02, 31.8, 32.1, 33.3, 34.8, 36.3, 37.1, 47.3, 48.6, 50.3, 112.6, 113.1, 113.2, 120.2, 130.8, 131.0, 132.4, 141.3, 142.0, 142.4, 166.9, 170.4, 170.6, 173.9. IR (KBr) cm−1: 2928, 2857, 1635, 1395. MS (ESI-TOF) m/z: 462 [M + Na]+. HR-MS (ESI-TOF) m/z: 462.1958 calcd for C22H34N3O2S35ClNa; Found: 462.1936.
tert-Butyl (4-(Methoxy(methyl)amino)-4-oxobutyl)carbamate (51)HOBt (186 mg, 1.4 mmol, 1.4 equiv.), MeONHMe·HCl (137 mg, 1.4 mmol, 1.4 equiv.), Et3N (548 mL, 3.9 mmol, 4.0 equiv.), and EDCI·HCl (264 mg, 1.4 mmol, 1.4 equiv.) were added to a solution of 50 (200 mg, 0.98 mmol) in CH2Cl2 (5 mL), and the mixture was stirred at r.t. for 12 h. Then, the reaction was quenched by the addition of sat. NaHCO3 aq. The whole mixture was extracted with AcOEt, and the AcOEt extract was washed with sat. NaHCO3 aq., and brine. Concentration of the AcOEt extract in vacuo gave a crude product which was purified by SiO2 column chromatography (hexane : AcOEt = 1 : 1) to give 51 (216 mg, 89%) as a colorless oil.
1H-NMR (600 MHz, CDCl3) δ: 1.40 (3H, s), 1.79 (2H, m), 2.44 (2H, s), 2.95–3.30 (5H, m), 3.65 (3H, s), 4.76 (1H, s). 13C-NMR (150 MHz, CDCl3) δ: 24.9, 28.5, 29.3, 32.3, 40.4, 61.3, 79.2, 156.1, 174.2. IR (KBr) cm−1: 3347 (br), 2975, 1711, 1523, 1173. MS (ESI-TOF) m/z: 269 [M + Na]+. HR-MS (ESI-TOF) m/z: 269.1477 calcd for C11H22N2O4Na; Found: 269.1464.
tert-Butyl (4-(Methoxy(methyl)amino)-4-oxobutyl)(methyl)carbamate (52)NaH (about 60% oil suspension, 37 mg, 0.93 mmol, 1.5 equiv.) was added to a solution of 51 (154 mg, 0.62 mmol) in DMF (3 mL) at 0°C, and the mixture was stirred at 0°C for 15 min. Iodomethane (388 µL, 6.2 mmol, 10 equiv.) was added to the amide solution, and the resulting mixture was stirred at r.t. for 12 h. Then, the reaction was quenched by the addition of sat. NH4Cl aq. The whole mixture was extracted with AcOEt, and the AcOEt extract was washed with NH4Cl aq. and brine. Concentration of the AcOEt extract in vacuo gave a crude product which was purified by SiO2 column chromatography (hexane : AcOEt = 1 : 1) to give 52 (148 mg, 91%) as a colorless oil.
1H-NMR (600 MHz, CDCl3) δ: 1.41 (9H, s), 1.71–1.87 (2H, m), 2.38 (2H, s), 2.81 (3H, s), 3.13 (3H, s), 3.23 (2H, t, J = 6.9 Hz), 3.63 (3H, s). 13C-NMR (150 MHz, CDCl3) δ: 22.7, 28.5, 28.9, 32.2, 34.1, 48.3, 61.2, 79.3, 155.9, 174.1. IR (KBr) cm−1: 2974, 1693, 1392, 1172. MS (ESI-TOF) m/z: 283 [M + Na]+. HR-MS (ESI-TOF) m/z: 283.1634 calcd for C12H24N2O4Na; Found: 283.1631.
tert-Butyl Methyl(4-oxopentyl)carbamate (53)CH3Li (3.0 M in Et2O, 128 µL, 0.38 mmol, 1.1 equiv.) was added to a solution of 52 (91 mg, 0.35 mmol) in Et2O (1 mL) at 0°C, and the mixture was stirred at 0°C for 0.5 h. Then, the reaction was quenched by the addition of sat. NH4Cl aq. The whole mixture was extracted with AcOEt, and the AcOEt extract was washed with NH4Cl aq. and brine. Concentration of the AcOEt extract in vacuo gave a crude product which was purified by SiO2 column chromatography (hexane : AcOEt = 3 : 1) to give 53 (56 mg, 75%) as a colorless oil. The spectroscopic and physical data were identical to those reported.11)
tert-Butyl (5-Chloro-4-methylpent-4-en-1-yl)(methyl)carbamate (54)LHMDS solution (1.0 M in THF, 436 µL, 0.44 mmol, 2.9 equiv.) was added to a solution of (chloromethyl)triphenylphosphonium chloride (156 mg, 0.45 mmol, 3.0 equiv.) in anhydrous THF (1 mL), and the mixture was stirred at r.t. for 15 min. A solution of 53 (32 mg, 0.15 mmol) in anhydrous THF (0.5 mL) was added dropwise to the ylide solution via cannula at r.t., and the resulting mixture was stirred at r.t. for 0.5 h. Then, the reaction was quenched by the addition of sat. NH4Cl aq. The whole mixture was extracted with AcOEt, and the AcOEt extract was washed with NH4Cl aq. and brine. Concentration of the AcOEt extract in vacuo gave a crude product which was purified by SiO2 column chromatography (hexane : AcOEt = 8 : 1) to give 54 (40 mg, quant.) as a colorless oil.
1H-NMR (600 MHz, CDCl3) δ: 1.40 (9H, s), 1.58 (2H, quint, J = 7.6 Hz), 1.67–1.77 (3H, m), 1.99 (1H, t, J = 7.6 Hz), 2.09–2.20 (1H, t, J = 7.6 Hz), 2.79 (3H, br s), 3.13 (2H, br s), 5.68–5.80 (1H, m). 13C-NMR (150 MHz, CDCl3) δ: 16.4, 20.8, 24.7, 25.1, 25.6, 25.7, 28.3, 28.5, 29.1, 34.2, 48.1, 48.2, 48.4, 48.8, 79.2, 79.3, 112.0, 112.3, 137.9, 155.7. IR (KBr) cm−1: 2975, 1696, 1393, 1166. MS (ESI-TOF) m/z: 270 [M + Na]+. HR-MS (ESI-TOF) m/z: 270.1237 calcd for C12H22NO235ClNa; Found: 270.1249.
(E)-N-((E)-5-Chloro-4-methylpent-4-en-1-yl)-3-methoxy-N-methylbut-2-enamide (55)TFA (120 µL, 1.6 mmol, 10 equiv.) was added to a solution of 54 (40 mg, 0.16 mmol) in CH2Cl2 (1 mL), and the mixture was stirred at r.t. for 2.5 h. Then, the reaction was quenched by the addition of sat. NaHCO3 aq. The whole mixture was extracted with AcOEt, and the AcOEt extract was washed with sat. NaHCO3 aq. and brine. Concentration of the AcOEt extract in vacuo gave an amine, which was used in the next reaction without further purification. The above crude product was dissolved to CH2Cl2 (1 mL). HOBt (32 mg, 0.24 mmol, 1.5 equiv.), 21 (28 mg, 0.24 mmol, 1.5 equiv.), Et3N (180 mL, 1.3 mmol, 8.0 equiv.), and EDCI·HCl (46 mg, 0.24 mmol, 1.5 equiv.) were added to the solution, and the resulting mixture was stirred at r.t. for 1 h. Then, the reaction was quenched by the addition of sat. NaHCO3 aq. The whole mixture was extracted with AcOEt, and the AcOEt extract was washed with sat. NaHCO3 aq., and brine. Concentration of the AcOEt extract in vacuo gave a crude product which was purified by SiO2 column chromatography (hexane : AcOEt = 2 : 1) to give 55 and 56 (9.7 mg, 25%, two steps) as a colorless oil.
55: 1H-NMR (600 MHz, CDCl3) δ: 1.68 (2H, m), 1.77 (3H, s), 2.06 (2H, t, J = 7.6 Hz), 2.18 (3H, s), 2.85–2.99 (3H, m), 3.32 (2H, m), 3.59 (3H, s), 5.14 (1H, s), 5.81 (1H, s). 13C-NMR (150 MHz, CDCl3) δ: 16.5, 18.9, 25.5, 26.0, 33.5, 34.0, 34.6, 36.1, 47.4, 49.7, 55.0, 91.0, 91.3, 112.3, 112.9, 137.5, 138.2, 168.2, 168.3, 168.7. IR (KBr) cm−1: 2932, 1650, 1380, 1240, 1158. MS (ESI-TOF) m/z: 268 [M + Na]+. HR-MS (ESI-TOF) m/z: 268.1080 calcd for C12H20NO235ClNa; Found: 268.1088.
(E)-2-Methylhept-2-enoic Acid (58)LiOH (83 mg, 3.5 mmol, 2.0 equiv.) was added to a solution of 57 (295 mg, 1.7 mmol) in THF : MeOH : H2O (1 : 1 : 1, 9 mL), and the mixture was stirred at r.t. for 20 h. The reaction was quenched by the addition of 5% HCl, and the whole mixture was extracted with AcOEt. Then, the AcOEt extract was washed with brine. Concentration of the AcOEt extract in vacuo gave a crude product which was purified by SiO2 column chromatography (hexane : AcOEt = 5 : 1) to give 58 (208 mg, 84%) as a colorless oil.
1H-NMR (600 MHz, CDCl3) δ: 0.87 (3H, t, J = 7.3 Hz), 1.25–1.44 (4H, m), 1.79 (3H, s), 2.11–2.20 (2H, m), 6.89 (1H, t, J = 7.6 Hz), 12.62 (1H, s). 13C-NMR (150 MHz, CDCl3) δ: 11.8, 13.7, 22.5, 28.6, 30.6, 127.1, 145.3, 174.1. IR (KBr) cm−1: 2959 (br), 1686, 1281. In mass mesurement, the spectra of sodium salt of compound 78 was obtained. MS (ESI-TOF) m/z: 187 [M + Na]+. HR-MS (ESI-TOF) m/z: 187.0711 calcd for C8H13O2Na2; Found: 187.0715.
(E)-N,2-Dimethyl-N-(thiazol-2-ylmethyl)hept-2-enamide (59)HOBt (85 mg, 0.63 mmol, 1.4 equiv.), amine 20 (114 mg, 0.89 mmol, 2.0 equiv.), Et3N (523 µL, 3.8 mmol, 6.0 equiv.), and EDCI·HCl (120 mg, 0.63 mmol, 1.4 equiv.) were added to a solution of 58 (64 mg, 0.45 mmol) in CH2Cl2 (5 mL), and the mixture was stirred at r.t. for 24 h. Then, the reaction was quenched by the addition of sat. NaHCO3 aq. The whole mixture was extracted with AcOEt, and the AcOEt extract was washed with sat. NaHCO3 aq., and brine. Concentration of the AcOEt extract in vacuo gave a crude product which was purified by SiO2 column chromatography (hexane : AcOEt = 1 : 1) to give 59 (103 mg, 91%) as a colorless oil.
1H-NMR (600 MHz, CDCl3) δ: 0.81 (3H, t, J = 6.8 Hz), 1.24 (2H, br s), 1.30 (2H, br s), 1.79 (3H, s), 2.03 (2H, dd, J = 14.0, 6.9 Hz), 2.97 (3H, s), 4.81 (2H, br s), 5.58 (1H, s), 7.26 (1H, br s), 7.64 (1H, br s). 13C-NMR (150 MHz, CDCl3) δ: 13.7, 14.0, 22.1, 27.1, 30.7, 36.1, 47.8, 119.9, 129.8, 130.3, 132.2, 141.9, 166.6, 173.6. IR (KBr) cm−1: 2927, 1631, 1393, 1074. MS (ESI-TOF) m/z: 275 [M + Na]+. HR-MS (ESI-TOF) m/z: 275.1194 calcd for C13H20N2OSNa; Found: 275.1201.