Experimental
All reagents and solvents were obtained from commercial sources and used as received. 1H-NMR spectra were recorded with tetramethylsilane as an internal standard using a JEOL JNM-Ex 270 MHz spectrometer. Automated column chromatographic separations were performed on a flash chromatography (Biotage ZIP®; Biotage, United Kingdom and Amino Inject Column; Yamazen, Osaka, Japan). LC/MS analysis was performed on a Waters Aquity Ultra Performance LC (Waters, United States) with a 2.1 × 50 mm Waters Aquity UPLC BEH C18 1.7 µm column. The column temperature was 40°C, with a run time of 2 min, flow rate of 0.6 mL/min, and a mixture of acetonitrile and water containing 0.1% trifluoroacetic acid with a gradient of 10–90% as an eluting solvent. The mass spectrometry data were acquired on a SQD2 quadrupole mass spectrometer (Waters).
Mixture of 4-{5-Iodo-1-[(2-(trimethylsilyl)ethoxy)methyl]-1H-pyrazol-4-yl}-2-(methylthio)pyrimidine and 4-{3-Iodo-1-[(2-(trimethylsilyl)ethoxy)methyl]-1H-pyrazol-4-yl}-2-(methylthio)pyrimidine (2)To a solution of 4-(5-iodo-1H-pyrazole-4yl)-2-(methylthio)pyrimidine 1 (2.01 g, 6.32 mmol) in N,N-dimethylformamide (DMF) (8 mL) was added Cs2CO3 (3.19 g, 9.79 mmol), and the reaction mixture was stirred at room temperature for 1 h. Then, 2-(trimethylsilyl)ethoxymethyl chloride (1.68 mL, 1.60 g, 9.60 mmol) was added slowly, and the reaction mixture was stirred at room temperature for 2 h. The reaction mixture was diluted with EtOAc, washed with brine, dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by column chromatography over silica gel (n-hexane–EtOAc) to afford the regioisomers 2 (2.47 g, 87%) as clear oil. 1H-NMR (270 MHz, CDCl3, major peaks of regioisomers) δ: 8.53 (1H, d, J = 4.9 Hz), 8.15 (1H, s), 7.66 (1H, d, J = 5.4 Hz), 5.46 (2H, s), 3.61–3.67 (2H, m), 2.63 (3H, s), 0.91–0.97 (2H, m), 0.01 (9H, s). LC/MS (electrospray ionization (ESI)): m/z 449 [M + H]+.
Mixture of 4-{5-Iodo-1-[(2-(trimethylsilyl)ethoxy)methyl]-1H-pyrazol-4-yl}-2-(methylsulfonyl)pyrimidine and 4-{3-Iodo-1-[(2-(trimethylsilyl)ethoxy)methyl]-1H-pyrazol-4-yl}-2-(methylsulfonyl)pyrimidine (3)To a solution of regioisomers 2 (2.47 g, 5.51 mmol) in tetrahydrofuran (THF) (40 mL) was added m-chloroperbenzoic acid (mCPBA) (3.17 g, 75%, 13.78 mmol) at 0°C. The reaction mixture was warmed to room temperature, stirred for 17 h, and concentrated in vacuo. The residue was purified twice by column chromatography over silica gel (n-hexane–EtOAc) to afford a mixture of regioisomers 3 (2.63 g, 99%) as a white powder. 1H-NMR (270 MHz, CDCl3, major peaks of regioisomers) δ: 8.88 (1H, d, J = 5.1 Hz), 8.33 (1H, s), 8.26 (1H, d, J = 5.4 Hz), 5.47 (2H, s), 3.61–3.67 (2H, t), 3.41 (3H, s), 0.91–0.96 (2H, t), 0.01 (9H, s). LC/MS (ESI): m/z 481 [M + H]+.
Mixture of 4-{5-Iodo-1-[(2-(trimethylsilyl)ethoxy)methyl]-1H-pyrazol-4-yl}-N-(4-methoxyphenyl)pyrimidin-2-amine and 4-{3-Iodo-1-[(2-(trimethylsilyl)ethoxy)methyl]-1H-pyrazol-4-yl}-N-(4-methoxyphenyl)pyrimidin-2-amine (4)A solution of compound 3 (2.62 g, 5.45 mmol) and p-anisidine (2.05 g, 16.65 mmol) in THF (20 mL) was refluxed for 17 h. The reaction mixture was cooled to room temperature, diluted with EtOAc, washed with brine, dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by column chromatography over silica gel (n-hexane–EtOAc) to afford a mixture of regioisomers 4 (1.79 g, 63%) as a beige solid. 1H-NMR (270 MHz, CDCl3, major peaks of regioisomers) δ: 8.41 (2H, d, J = 5.1 Hz), 8.05 (1H, s), 7.52 (1H, d, J = 9.2 Hz), 7.38 (1H, d, J = 5.1 Hz), 6.91 (1H, d, J = 8.9 Hz), 5.46 (2H, s), 3.82 (3H, s), 3.64 (2H, t), 0.94 (2H, t), 0.01 (9H, s). LC/MS (ESI): m/z 524 [M + H]+.
N-(4-Methoxyphenyl)-4-(5-(pyridin-3-yl)-1-[(2-(trimethylsilyl)ethoxy)methyl]-1H-pyrazol-4-yl)pyrimidin-2-amine and N-(4-Methoxyphenyl)-4-(3-(pyridin-3-yl)-1-[(2-(trimethylsilyl)ethoxy)methyl]-1H-pyrazol-4-yl)pyrimidin-2-amine (5a)A mixture of compound 4 (104.6 mg, 0.200 mmol), pyridine-3-boronic acid (52.1 mg, 0.424 mmol), Pd2(dba)3 (6.7 mg, 0.007 mmol), K3PO4 (198.2 mg, 0.934 mmol), H2O (0.5 mL), dioxane (0.5 mL), and 0.6 M tricyclohexylphosphine (20 µL, 0.012 mmol) was refluxed under an argon atmosphere for 15 h. The reaction mixture was filtered, washed with EtOAc, and concentrated in vacuo. The residue was purified by column chromatography over silica gel (n-hexane–EtOAc) to give a mixture of regioisomers 5a (40.2 mg, 43%) as a yellow oil. 1H-NMR (270 MHz, CDCl3, major peaks of regioisomers) δ: 8.86 (1H, d, J = 1.9 Hz), 8.62–8.64 (1H, m), 8.25 (1H, d, J = 5.4 Hz), 8.14 (1H, s), 7.88–7.91 (2H, m), 7.30–7.37 (2H, m), 7.10 (1H, s), 6.79–6.83 (2H, m), 6.56 (1H, d, J = 5.4 Hz), 5.51 (2H, s), 3.80 (3H, s), 3.67–3.80 (2H, s), 0.91–1.01 (2H, m), 0.01 (9H, s). LC/MS (ESI): m/z 475 [M + H]+.
N-(4-Methoxyphenyl)-4-(3-(pyridin-3-yl)-1H-pyrazol-4-yl)pyrimidin-2-amine (6a)To a solution of compound 5a (40 mg, 0.084 mmol) in dry THF (6 mL) was added 1 M TBAF in THF (0.17 µL). The reaction mixture was refluxed for 3.5 h and added to brine, extracted with EtOAc, dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by column chromatography over silica gel (CH2Cl2–MeOH) to afford compound 6a (23.3 mg, 81%) as a yellow solid. 1H-NMR (270 MHz, CD3OD) δ: 8.70–8.71 (1H, m), 8.49 (1H, dd, J = 1.6, 4.9 Hz), 8.26 (1H, s), 8.24 (1H, s), 7.99–8.03 (1H, m), 7.40–7.44 (1H, m), 7.22 (2H, d, J = 8.9 Hz), 6.82 (2H, d, J = 5.1 Hz), 6.67 (2H, d, J = 8.9 Hz), 3.76 (3H, s). LC/MS (ESI): m/z 345 [M + H]+.
Compounds 6c–e were prepared following a procedure similar to that used to prepare compound 6a.
N-(4-Methoxyphenyl)-4-(3-pyridin-4-yl-1H-pyrazol-4-yl)pyrimidin-2-amine (6c)1H-NMR (270 MHz, CD3OD) δ: 8.49–8.52 (2H, m), 8.25–8.28 (2H, m), 7.62–7.64 (2H, m), 7.26 (2H, d, J = 9.2 Hz), 6.83 (1H, d, J = 5.1 Hz), 6.68 (2H, d, J = 9.2 Hz), 3.75 (3H, s). LC/MS (ESI): m/z 345 [M + H]+.
N-(4-Methoxyphenyl)-4-(3-pyrimidin-5-yl-1H-pyrazol-4-yl)pyrimidin-2-amine (6d)1H-NMR (270 MHz, DMSO-d6) δ: 13.68 (1H, s), 9.17 (1H, s), 9.07 (1H, s), 8.90 (2H, s), 8.49 (1H, s), 8.35 (1H, d, J = 4.9 Hz), 7.20 (2H, d, J = 8.9 Hz), 6.93 (1H, d, J = 4.9 Hz), 6.58 (2H, d, J = 8.9 Hz), 3.67 (3H, s). LC/MS (ESI): m/z 346 [M + H]+.
N-(4-Methoxyphenyl)-4-(3-quinolin-3-yl-1H-pyrazol-4-yl)pyrimidin-2-amine (6e)1H-NMR (270 MHz, CD3OD) δ: 8.92 (1H, d, J = 2.4 Hz), 8.57 (1H, d, J = 1.6 Hz), 8.32 (1H, br s), 8.29 (1H, d, J = 8.3 Hz), 7.94–8.00 (2H, m), 7.77–7.80 (1H, m), 7.62–7.68 (1H, m), 6.95–6.98 (3H, m), 6.13 (2H, d, J = 8.9 Hz), 3.49 (3H, s). LC/MS (ESI): m/z 395 [M + H]+.
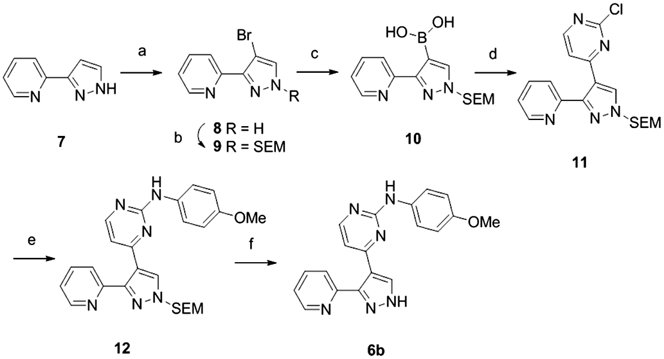
2-(4-Bromo-1H-pyrazol-3-yl)pyridine (8)A solution of 2-(1H-pyrazole-3-yl) pyridine (3.02 g, 20.80 mmol) and NBS (3.72 g, 20.90 mmol) in DMF (30 mL) was stirred for 2 h in an ice bath. The reaction mixture was poured into ice water, filtered, and dried in vacuo. The residue (4.38 g, 95% yield) as a white solid was reacted without further purification. 1H-NMR (270 MHz, CDCl3) δ: 11.46 (1H, br s), 8.63–8.65 (1H, m), 8.29 (1H, d, J = 8.4 Hz), 7.78–7.85 (1H, m), 7.64 (1H, s), 7.28–7.33 (1H, m). LC/MS (ESI): m/z 223 [M + H]+.
2-{4-Bromo-1-[(2-(trimethylsilyl)ethoxy)methyl]-1H-pyrazol-3-yl}pyridine (9)To a solution of compound 8 (4.38 g, 19.55 mmol) and Cs2CO3 (14.01 g, 43.00 mmol) in DMF (35 mL) was added 2-(trimethylsilyl)ethoxymethyl chloride (7.27 mL, 43.01 mmol) at room temperature for 1 h. The reaction mixture was diluted with EtOAc, washed with brine, dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by column chromatography over silica gel (n-hexane–EtOAc) to give compound 9 as a yellow oil (3.23 g, 47%). 1H-NMR (270 MHz, CDCl3) δ: 8.73–8.76 (1H, m), 7.98–8.01 (1H, m), 7.73–7.80 (1H, m), 7.30 (1H, s), 7.27–7.30 (1H, m), 5.48 (2H, s), 3.59–3.65 (2H, m), 0.90–1.00 (2H, m), −0.03 (9H, s). LC/MS (ESI): m/z 354 [M + H]+.
{3-(Pyridin-2-yl)-1-[(2-(trimethylsilyl)ethoxy)methyl]-1H-pyrazol-4-yl}boronic Acid (10)To a solution of compound 9 (3.23 g, 9.12 mmol) and triisoppropyl borate (5.47 mL, 23.71 mmol) in THF (50 mL) was added n-BuLi (1.4 M solution in hexane, 8.42 mL, 21.89 mmol) at −78°C. After stirring for 1 h, the reaction mixture was warmed to 0°C for 1 h, quenched with saturated NH4Cl aqueous solution, and extracted with CH2Cl2. The extract was dried over Na2SO4 and concentrated in vacuo. The residue was purified by column chromatography over silica gel (hexane–EtOAc) to afford compound 10 (1.92 g, 66% yield) as a white solid. 1H-NMR (270 MHz, CDCl3) δ: 8.74 (2H, br s), 8.52–8.55 (1H, m), 8.32–8.36 (1H, m), 8.01 (1H, m), 7.79–7.86 (1H, m), 7.27–7.32 (1H, m), 5.48 (2H, s), 3.60–3.66 (2H, m), 0.91–0.98 (2H, m), −0.12 (9H, s). LC/MS (ESI): m/z 320 [M + H]+.
2-Chloro-4-{3-(pyridin-2-yl)-1-[(2-(trimethylsilyl)ethoxy)methyl]-1H-pyrazol-4-yl}pyrimidine (11)To a solution of compound 10 (0.51 g, 1.60 mmol), 2,4-dichloropyrimidine (0.29 g, 1.95 mmol), and 2N Na2CO3 aq (1.4 mL) in DME–EtOH (3.0 : 6.0 mL) was added Pd(PPh3)2Cl2 (0.08 g, 0.11 mmol). After the replacement of nitrogen, the reaction mixture was heated at 80°C for 4.5 h. The reaction mixture was added to water, extracted with EtOAc, dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by column chromatography over silica gel (n-hexane–EtOAc) to afford compound 11 as a yellow oil (0.60 g, 96% yield). 1H-NMR (270 MHz, CDCl3) δ: 8.70–8.73 (1H, m), 8.49 (2H, d, J = 5.7 Hz), 7.83–7.93 (2H, m), 7.76 (1H, d, J = 5.4 Hz), 7.37–7.45 (1H, m), 5.57 (2H, s), 3.70–3.76 (2H, m), 0.99–1.05 (2H, m), 0.52 (9H, s). LC/MS (ESI): m/z 388 [M + H]+.
N-(4-Methoxyphenyl)-4-{3-(pyridin-2-yl)-1-[2-(trimethylsilyl)ethoxy)methyl]-1H-pyrazol-4-yl}pyrimidin-2-amine (12)The title compound 12 was prepared from compound 11 and p-anisidine in a procedure similar to that used to prepare compound 4. 1H-NMR (270 MHz, CDCl3) δ: 8.68–8.70 (1H, m), 8.24 (1H, d, J = 5.1 Hz), 8.20 (1H, s), 7.69–7.79 (2H, m), 7.39–7.44 (2H, m), 7.28–7.32 (1H, m), 6.90 (1H, s), 6.81–6.87 (3H, m), 5.53 (2H, s), 3.80 (3H, s), 3.66–3.72 (2H, m), 0.94–1.00 (2H, m), 0.12 (9H, s). LC/MS (ESI): m/z 475 [M + H]+.
N-(4-Methoxyphenyl)-4-(3-(pyridin-2-yl-1H-pyrazol-4-yl)pyrimidin-2-amine (6b)The title compound 6b was prepared from SEM-protected 12 following a procedure similar to that used to prepare compound 6a. 1H-NMR (270 MHz, CD3OD) δ: 8.54–8.63 (1H, m), 8.06–8.30 (2H, m), 7.72–7.89 (2H, m), 7.28–7.40 (3H, m), 6.60–6.85 (3H, m), 3.76 (3H, s). LC/MS (ESI): m/z 345 [M + H]+.
2-{4-[2-(4-Methoxyanilino)pyrimidin-4-yl]-3-pyridin-3-ylpyrazol-1-yl}ethanol (13a)To a solution of compound 6a (52.5 mg, 0.152 mmol) and K2CO3 (53.2 mg, 0.385 mmol) in DMF (0.4 mL) was added 2-iodoethanol (87.2 g, 0.507 mmol) at 60°C overnight. The reaction mixture was poured into water and extracted with EtOAc. The extract was dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by column chromatography over amino silica gel (CH2Cl2–MeOH) to afford the N1-isomer (1.7 mg, 3% yield) and the N2-regioisomer 13a (4.2 mg, 7% yield) as a yellow solid. 1H-NMR (270 MHz, CDCl3) δ: 8.83–8.84 (1H, m), 8.60–8.63 (1H, m), 8.23 (1H, d, J = 5.1 Hz), 8.03 (1H, s), 7.88–7.91 (1H, m), 7.34–7.37 (2H, m), 7.31–7.32 (1H, m), 6.93 (1H, br s), 6.80–6.85 (2H, m), 6.55 (1H, d, J = 5.1 Hz), 4.34–4.37 (2H, m), 4.10–4.13 (2H, m), 3.80 (3H, s). LC/MS (ESI) 389 [M + H]+.
Compounds 13b–e were prepared following a procedure similar to that used to prepare compound 13a.
4-[1-(2-Methoxyethyl)-3-pyridin-3-ylpyrazol-4-yl]-N-(4-methoxyphenyl)pyrimidin-2-amine (13b)1H-NMR (270 MHz, CDCl3) δ: 8.85 (1H, d, J = 1.9 Hz), 8.60–8.62 (1H, m), 8.22 (1H, d, J = 5.1 Hz), 8.05 (1H, s), 7.87–7.91 (1H, m), 7.35 (2H, dd, J = 6.8, 2.2 Hz), 7.28–7.31 (1H, m), 6.89 (1H, s), 6.81 (2H, dd, J = 6.8, 2.2 Hz), 6.56 (1H, d, J = 5.1 Hz), 4.38 (2H, t, J = 5.1 Hz), 3.83 (2H, t, J = 5.1 Hz), 3.80 (3H, s), 3.40 (3H, s). LC/MS (ESI): m/z 403 [M + H]+.
N-(4-Methoxyphenyl)-4-[1-(oxetan-3-yl)-3-pyridin-3-ylpyrazol-4-yl]pyrimidin-2-amine (13c)1H-NMR (270 MHz, CDCl3) δ: 8.86 (1H, d, J = 1.6 Hz), 8.62–8.64 (1H, m), 8.24 (1H, d, J = 5.4 Hz), 8.15 (1H, s), 7.90–7.94 (1H, m), 7.31–7.38 (3H, m), 6.92 (1H, s), 6.81–6.85 (2H, m), 6.55 (1H, d, J = 5.1 Hz), 5.50–5.60 (1H, m), 5.08–5.19 (4H, m), 3.80 (3H, s). LC/MS (ESI): m/z 401 [M + H]+.
N-(4-Methoxyphenyl)-4-[3-pyridin-3-yl-1-(2,2,2-trifluoroethyl)pyrazol-4-yl]pyrimidin-2-amine (13d)1H-NMR (270 MHz, CDCl3) δ: 8.83–8.84 (1H, m), 8.64 (1H, d, J = 4.9, 1.6 Hz), 8.26 (1H, d, J = 5.4 Hz), 8.08 (1H, s), 7.88–7.92 (1H, m), 7.31–7.36 (3H, m), 6.91 (1H, s), 6.79–6.85 (2H, m), 6.55 (1H, d, J = 5.1 Hz), 4.81 (2H, m), 3.80 (3H, s). LC/MS (ESI): m/z 427 [M + H]+.
4-(1-Ethyl-5-pyridin-3-ylpyrazol-4-yl)-N-(4-methoxyphenyl)pyrimidin-2-amine (13e)1H-NMR (270 MHz, CDCl3) δ: 9.36 (1H, d, J = 5.7 Hz), 8.60–8.73 (3H, s), 8.21 (1H, d, J = 5.1 Hz), 7.92–7.99 (3H, m), 7.07–7.11 (2H, m), 6.73 (1H, d, J = 5.1 Hz), 5.83 (1H, br s), 4.09–4.17 (2H, m), 3.87 (3H, s), 1.40 (3H, t, J = 7.0 Hz). LC/MS (ESI): m/z 373 [M + H]+.
4-(1-Ethyl-3-pyridin-3-ylpyrazol-4-yl)-N-(4-methoxyphenyl)pyrimidin-2-amine (13f)1H-NMR (270 MHz, CDCl3) δ: 8.84–8.85 (1H, m), 8.61 (1H, d, J = 5.1, 1.6 Hz), 8.21 (1H, d, J = 5.4 Hz), 7.98 (1H, s), 7.87–7.92 (1H, m), 7.33–7.39 (2H, m), 7.29–7.32 (1H, m), 6.93 (1H, s), 6.79–6.85 (2H, m), 6.53 (1H, d, J = 4.9 Hz), 4.27 (2H, q, J = 7.3 Hz), 3.80 (3H, s), 1.59 (3H, t, J = 7.3 Hz). LC/MS (ESI): m/z 373 [M + H]+.
N-(4-Methoxyphenyl)-4-(1-propan-2-yl-5-pyridin-3-ylpyrazol-4-yl)pyrimidin-2-amine (13g)1H-NMR (270 MHz, CDCl3) δ: 8.74 (1H, dd, J = 4.9, 1.6 Hz), 8.66 (1H, d, J = 2.1 Hz), 8.16 (1H, s), 8.14 (1H, d, J = 5.4 Hz), 7.67–7.71 (1H, m), 7.40–7.44 (1H, m), 7.28–7.31 (2H, m), 6.91 (1H, br s), 6.80–6.83 (2H, m), 6.33 (1H, d, J = 5.4 Hz), 4.24–4.34 (1H, m), 3.79 (3H, s), 1.49 (3H, s), 1.46 (3H, s). LC/MS (ESI): m/z 387 [M + H]+.
N-(4-Methoxyphenyl)-4-(1-propan-2-yl-3-pyridin-3-ylpyrazol-4-yl)pyrimidin-2-amine (13h)1H-NMR (270 MHz, CDCl3) δ: 8.85 (1H, d, J = 1.4 Hz), 8.61 (1H, d, J = 3.2 Hz), 8.21 (1H, d, J = 5.4 Hz), 8.00 (1H, s), 7.88–7.92 (1H, m), 7.33–7.39 (2H, d, J = 8.9 Hz), 7.29–7.32 (1H, m), 7.02 (1H, br s), 6.82 (2H, d, J = 8.9 Hz), 6.54 (1H, d, J = 5.1 Hz), 4.54–4.64 (1H, m), 3.80 (3H, s), 1.61 (3H, s), 1.59 (3H, s). LC/MS (ESI): m/z 387 [M + H]+.
2-(2-Chloropyrimidin-4-yl)-1-(pyridin-3-yl)ethanone (15)To a solution of methyl nicotinate 14 (26.6 g, 0.194 mol), 2-chloro-4-methylpyridine (25.0 g, 0.194 mol) in THF (250 mL) at −30°C was added LiHMDS (0.389 mol) dropwise. The mixture was stirred for 40 min at −30°C and then for 2 h at room temperature. The solution was diluted with saturated NH4Cl aqueous solution, and the aqueous phase was extracted with EtOAc. The extracts were dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by chromatography over silica gel (EtOAc). The resulting solid was washed with MTBE to afford compound 15 (30.6 g, 68% yield) as an orange solid. LC/MS (ESI): m/z 234 [M + H]+.
2-Chloro-4-(3-(pyridin-3-yl)-1H-pyrazol-4-yl)pyrimidine (16)A solution of compound 15 (30.6 g, 0.131 mol), N,N-dimethylformamide dimethyl acetal (18.7 g, 0.157 mol), and AcOH (9.44 g, 0.157 mol) in toluene (459 mL) was heated at 110°C for 1.5 h. N,N-dimethylformamide dimethyl acetal (3.74 g, 0.031 mol) was added to the mixture, and it was stirred at 110°C for 1 h. After cooling to 45°C, the mixture was concentrated in vacuo. To a solution of the residue in EtOH (245 mL), AcOH (9.44 g) and hydrazine monohydrate (6.56 g, 0.131 mol) were added and stirred in an ice bath for 75 min. Then ice water was added, filtered, and concentrated in vacuo to afford 16 (19.4 g, 58% yield). 1H-NMR (270 MHz, CDCl3) δ: 8.82 (1H, d, J = 2.3 Hz), 8.72 (1H, dd, J = 4.6, 1.6 Hz), 8.42 (1H, d, J = 5.3 Hz), 8.33 (1H, s), 7.90–7.98 (1H, m), 7.39–7.48 (1H, m), 7.07 (1H, d, J = 5.3 Hz). LC/MS (ESI): m/z 258 [M + H]+
2-Chloro-4-(1-ethyl-3-(pyridin-3-yl)-1H-pyrazol-4-yl)pyrimidine (17)To a solution of compound 16 (9.17 g, 35.6 mmol) and K2CO3 (7.38 g, 53.4 mmol) in DMF (73 mL) was added ethyl iodide (6.10 g, 39.1 mmol) at room temperature for 6 h. The mixture was poured into water, and the aqueous phase was extracted with EtOAc. The extracts were dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by column chromatography over silica gel (CH3Cl–MeOH to CH3Cl–MeOH). The resulting solid was washed with MTBE to afford compound 17 (4.30 g, 42% yield) as a yellow solid. 1H-NMR (270 MHz, CDCl3) δ: 8.79 (1H, dd, J = 2.1, 0.8 Hz), 8.68 (1H, dd, J = 4.9, 1.6 Hz), 8.36 (1H, d, J = 5.3 Hz), 8.22 (1H, s), 7.89 (1H, dt, J = 7.7, 2.1 Hz), 7.26–7.41 (1H, ddd, J = 7.8, 4.9, 0.8 Hz), 6.96 (1H, d, J = 5.2 Hz), 4.28 (2H, q, J = 7.3 Hz), 1.58–1.62 (overlapped by a water signal, 5H, m). LC/MS (ESI): m/z 286 [M + H]+.
4-(1-Ethyl-3-(pyridin-3-yl)-1H-pyrazol-4-yl)-2-(4-methoxybenzyl)pyrimidine (18a)To a solution of compound 17 (150 mg, 0.525 mmol) and Pd(PPh3)4 (30 mg, 0.026 mmol) in THF (5 mL) was added 4-(methoxybenzoyl) zinc chloride (2.0 mL) dropwise at room temperature. The mixture was stirred for 8 h at 60°C. The solution was poured into saturated NH4Cl aqueous solution and Na2 ethylenediaminetetraacetic acid (EDTA), and the aqueous phase was extracted with EtOAc. The extract was dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by chromatography over silica gel (CH2Cl2–MeOH) to afford compound 18a (74.25 mg, 38% yield) as a clear oil. 1H-NMR (270 MHz, CDCl3) δ: 8.81–8.82 (1H, m), 8.62–8.65 (1H, m), 8.45 (1H, d, J = 5.4 Hz), 8.06 (1H, s), 7.85–7.89 (1H, m), 7.27–7.34 (2H, m), 7.23–7.25 (2H, m), 6.81–6.91 (3H, m), 4.28 (2H, m), 4.16 (2H, s), 3.78 (3H, s), 1.58 (3H, t), 1.57–1.60 (overlapped by a water signal, 5.8H, m). LC/MS (ESI): m/z 372 [M + H]+.
4-(1-Ethyl-3-pyridin-3-ylpyrazol-4-yl)-N-(3-methoxyphenyl)pyrimidin-2-amine (18b)A solution of compound 17 (50 mg, 0.175 mmol) and m-anisidine (19.7 µL, 21.6 mg, 0.175 mmol) in MeOH (0.5 mL) was heated for 22 h in a sealed tube. After cooling to room temperature, the mixture was concentrated in vacuo. The residue was purified by column chromatography over amino silica gel (CH2Cl2–MeOH) to afford compound 18b (35 mg, 53% yield) as a beige solid. 1H-NMR (270 MHz, CDCl3) δ: 8.83–8.84 (1H, m), 8.61–8.63 (1H, m), 8.23 (1H, d, J = 5.1 Hz), 8.00 (1H, s), 7.87–7.92 (2H, m), 7.29–7.34 (1H, m), 7.14–7.21 (2H, m), 6.97–7.03 (1H, m), 6.77 (1H, s), 6.55 (1H, d, J = 5.1 Hz), 4.23–4.32 (2H, m), 2.32 (3H, s), 1.59 (3H, t). LC/MS (ESI): m/z 373 [M + H]+.
4-(1-Ethyl-3-pyridin-3-ylpyrazol-4-yl)-N-(2-methoxyphenyl)pyrimidin-2-amine (18c)The title compound 18c was prepared from compound 17 and o-anisidine following a procedure similar to that used to prepare compound 18b. 1H-NMR (270 MHz, CDCl3) δ: 8.85–8.86 (1H, m), 8.62 (1H, d, J = 1.6, 4.6 Hz), 8.23–8.28 (2H, m), 8.04 (1H, s), 7.88–7.92 (1H, m), 7.71 (1H, s), 7.30–7.35 (1H, m), 6.87–6.94 (3H, m), 6.56 (1H, d, J = 5.4 Hz), 4.25–4.33 (2H, m), 1.61 (3H, t, J = 7.3 Hz). LC/MS (ESI): m/z 373 [M + H]+.
N-[4-(Difluoromethoxy)phenyl]-4-(1-ethyl-3-pyridin-3-ylpyrazol-4-yl)pyrimidin-2-amine (18d)The title compound 18d was prepared from compound 17 and 4-(difluoromethoxy)aniline following a procedure similar to that used to prepare compound 18b. 1H-NMR (270 MHz, CDCl3) δ: 8.83 (1H, d, J = 1.1 Hz), 8.61–8.62 (1H, m), 8.26 (1H, d, J = 2.7 Hz), 7.99 (1H, s), 7.90 (1H, d, J = 4.3 Hz), 7.46 (2H, d, J = 4.6 Hz), 7.29–7.34 (1H, m), 7.01–7.23 (3H, m), 6.31–6.62 (2H, m), 4.27–4.31 (2H, m), 1.57–1.63 (overlapped by a water signal, 15H, t). LC/MS (ESI): m/z 409 [M + H]+.
4-(1-Ethyl-3-pyridin-3-ylpyrazol-4-yl)-N-(4-piperidin-4-yloxyphenyl)pyrimidin-2-amine (18e)Boc-protected 18e, tert-butyl 4-{4-[(4-(1-ethyl-3-(pyridin-3-yl)-1H-pyrazol-4-yl)pyrimidin-2-yl)amino]phenoxy}piperidine-1-carboxylate, was prepared from compound 17 and tert-butyl 4-(4-aminophenoxy)piperidine-1-carboxylate following a procedure similar to that used to prepare compound 18b. To a solution of boc-protected 18e (170 mg, 0.314 mmol) in CH2Cl2 was added trifluoroacetic acid (607 µL, 904.4 mg, 7.932 mmol) in an ice bath. The mixture was warmed to room temperature and stirred for 16 h. The solution was added to saturated NaHCO3 aqueous solution, and the aqueous phase was extracted with CH2Cl2. The extract was washed with brine, dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by column chromatography over amino silica gel (CH2Cl2–MeOH) to afford compound 18e (104.8 mg, 75% yield) as a yellow solid. 1H-NMR (270 MHz, CDCl3) δ: 8.84–8.85 (1H, m), 8.60–8.62 (1H, m), 8.22 (1H, d, J = 5.1 Hz), 7.98 (1H, s), 7.87–7.92 (1H, m), 7.35 (2H, d, J = 8.6 Hz), 7.29–7.32 (1H, m), 7.07 (1H, s), 6.83 (2H, d, J = 8.9 Hz), 6.54 (1H, d, J = 5.1 Hz), 4.23–4.36 (3H, m), 3.12–3.19 (2H, m), 2.71–2.80 (2H, m), 2.20 (1H, br s), 1.99–2.06 (2H, m), 1.66–1.72 (2H, m), 1.59 (3H, t). LC/MS (ESI): m/z 442 [M + H]+.
4-(1-Ethyl-3-pyridin-3-ylpyrazol-4-yl)-N-(4-piperidin-4-ylphenyl)pyrimidin-2-amine (18f)The title compound 18f was prepared from compound 17 and tert-butyl 4-(4-aminophenyl)-1-piperidinecarboxylate following a procedure similar to that used to prepare compound 18e. 1H-NMR (270 MHz, CDCl3) δ: 8.77–8.88 (1H, m), 8.54 (1H, dd, J = 4.6, 1.6 Hz), 8.17 (1H, d, J = 4.9 Hz), 7.93 (1H, s), 7.80–7.85 (1H, m), 7.34 (2H, d, J = 8.4 Hz), 7.23–7.27 (1H, m), 7.05 (2H, d, J = 8.6 Hz), 6.97 (1H, s), 6.49 (1H, d, J = 5.4 Hz), 4.21 (2H, q, J = 7.3 Hz), 3.14–3.18 (2H, m), 2.66–2.74 (2H, m), 2.49–2.56 (1H, m), 1.58–1.81 (4H, m), 1.53 (3H, t, J = 7.3 Hz). LC/MS (ESI): m/z 426 [M + H]+.
4-(1-Ethyl-3-pyridin-3-ylpyrazol-4-yl)-N-(4-piperazin-1-ylphenyl)pyrimidin-2-amine (18g)The title compound 18g was prepared from compound 17 and tert-butyl 4-(4-aminophenyl)piperazine-1-carboxylate following a procedure similar to that used to prepare compound 18e. 1H-NMR (270 MHz, CDCl3) δ: 8.55–8.86 (1H, m), 8.61–8.62 (1H, m), 8.21 (1H, d, J = 5.1 Hz), 7.98 (1H, s), 7.87–7.91 (1H, m), 7.35 (2H, d, J = 8.6 Hz), 7.30–7.31 (1H, m), 6.93 (1H, s), 6.86 (2H, d, J = 8.9 Hz), 6.51 (1H, d, J = 5.4 Hz), 4.23–4.31 (2H, m), 3.05–3.12 (8H, m), 1.58–1.63 (3H, m). LC/MS (ESI): m/z 427 [M + H]+.
4-(1-Ethyl-3-pyridin-3-ylpyrazol-4-yl)-N-(4-piperidin-1-ylphenyl)pyrimidin-2-amine (18h)The title compound 18h was prepared from compound 17 and 4-(1-piperidino)aniline following a procedure similar to that used to prepare compound 18b. 1H-NMR (270 MHz, CDCl3) δ: 8.85 (1H, dd, J = 2.2, 1.1 Hz), 8.60–8.62 (1H, m), 8.20 (1H, d, J = 5.4 Hz), 7.98 (1H, s), 7.87–7.91 (1H, m), 7.28–7.35 (3H, m), 6.86–6.91 (3H, m), 6.50 (1H, d, J = 5.1 Hz), 4.23–4.31 (2H, m), 3.07–3.11 (4H, m), 1.68–1.77 (overlapped by a water signal, 12H, m), 1.52–1.62 (5H, m). LC/MS (ESI): m/z 426 [M + H]+.
N-[4-(4-Aminopiperidin-1-yl)phenyl]-4-(1-ethyl-3-pyridin-3-ylpyrazol-4-yl)pyrimidin-2-amine (18i)The title compound 18i was synthesized from compound 17 and tert-butyl (1-(4-aminophenyl)piperidin-4-yl)carbamate following a procedure similar to that used to prepare compound 18e. 1H-NMR (270 MHz, CDCl3) δ: 8.83–8.87 (1H, m), 8.60 (1H, dd, J = 4.8, 1.8 Hz), 8.21 (1H, d, J = 5.3 Hz), 7.98 (1H, s), 7.89 (1H, dt, J = 7.9, 2.0 Hz), 7.28–7.45 (3H, m), 6.79–7.03 (3H, m), 6.51 (1H, d, J = 4.9 Hz), 4.27 (2H, q, J = 7.4 Hz), 3.50–3.62 (2H, m), 2.61–2.94 (3H, m), 1.87–1.98 (2H, m), 1.59 (3H, t, J = 7.4 Hz), 1.47–1.55 (2H, m). LC/MS (ESI): m/z 441 [M + H]+.
4-(1-Ethyl-3-pyridin-3-ylpyrazol-4-yl)-N-(3-piperazin-1-ylphenyl)pyrimidin-2-amine (18j)The title compound 18j was synthesized from compound 17 and tert-butyl 4-(3-aminophenyl)piperidine-1-carboxylate following a procedure similar to that used to prepare compound 18e. 1H-NMR (270 MHz, CDCl3) δ: 8.85–8.86 (1H, m), 8.60–8.63 (1H, m), 8.23 (1H, d, J = 5.4 Hz), 8.03 (1H, s), 7.88–7.92 (1H, m), 7.27–7.35 (3H, m), 7.16–7.20 (1H, m), 7.01–7.03 (1H, m), 6.54–6.63 (2H, m), 4.24–4.31 (2H, m), 3.14–3.18 (4H, m), 3.01–3.06 (4H, m), 1.57–1.62 (3H, m). LC/MS (ESI): m/z 427 [M + H]+.
4-(1-Ethyl-3-pyridin-3-ylpyrazol-4-yl)-N-(6-piperazin-1-ylpyridin-3-yl)pyrimidin-2-amine (18k)The title compound 18k was synthesized from compound 17 and tert-butyl 4-(5-aminopyridin-2-yl)piperazine-1-carboxylate following a procedure similar to that used to prepare compound 18e. 1H-NMR (270 MHz, CDCl3) δ: 8.83–8.84 (1H, m), 8.60–8.61 (1H, m), 8.26 (1H, d, J = 2.7 Hz), 8.20 (1H, d, J = 5.1 Hz), 7.98 (1H, s), 7.85–7.90 (1H, m), 7.71–7.74 (1H, m), 7.28–7.33 (1H, m), 6.87 (1H, s), 6.55–6.60 (2H, m), 4.27 (2H, q, J = 7.3 Hz), 3.65–3.69 (4H, m), 3.17–3.21 (4H, m), 1.57–1.62 (3H, m). LC/MS (ESI): m/z 428 [M + H]+.
4-(1-Ethyl-3-pyridin-3-ylpyrazol-4-yl)-N-(5-piperazin-1-ylpyridin-2-yl)pyrimidin-2-amine (18l)The title compound 18l was prepared from compound 17 and tert-butyl 4-(6-aminopyridin-3-yl)piperazine-1-carboxylate following a procedure similar to that used to prepare compound 18e. 1H-NMR (270 MHz, CDCl3) δ: 8.86–8.87 (1H, m), 8.59–8.61 (1H, m), 8.30 (1H, d, J = 5.1 Hz), 7.99 (1H, s), 7.84–7.89 (3H, m), 7.65 (1H, br s), 7.29–7.33 (1H, m), 7.11–7.15 (1H, m), 6.64 (1H, d, J = 5.4 Hz), 4.25–4.33 (2H, m), 3.04–3.11 (8H, m), 1.58–1.64 (overlapped by a water signal, 10H, m). LC/MS (ESI): m/z 428 [M + H]+.
N-(3-Chloro-4-piperazin-1-ylphenyl)-4-(1-ethyl-3-pyridin-3-ylpyrazol-4-yl)pyrimidin-2-amine (18m)The title compound 18m was prepared from compound 17 and tert-butyl 4-(4-amino-2-chlorophenyl)piperazine-1-carboxylate following a procedure similar to that used to prepare compound 18e. 1H-NMR (270 MHz, CDCl3) δ: 8.83–8.84 (1H, m), 8.61–8.63 (1H, m), 8.23 (1H, d, J = 5.1 Hz), 8.05 (1H, s), 7.88–7.92 (1H, m), 7.79 (1H, d, J = 2.4 Hz), 7.31–7.36 (1H, m), 7.21–7.31 (1H, m), 6.96 (2H, d, J = 8.6 Hz), 6.56 (1H, d, J = 4.9 Hz), 4.25–4.33 (2H, m), 2.98–3.08 (8H, m), 1.58–1.64 (3H, t). LC/MS (ESI): m/z 461 [M + H]+.
4-(1-Ethyl-3-pyridin-3-ylpyrazol-4-yl)-N-(3-methyl-4-piperazin-1-ylphenyl)pyrimidin-2-amine (18n)The title compound 18n was prepared from compound 17 and tert-butyl 4-(4-amino-2-methylphenyl)piperazine-1-carboxylate following a procedure similar to that used to prepare compound 18e. 1H-NMR (270 MHz, CDCl3) δ: 8.84–8.85 (1H, m), 8.60–8.62 (1H, m), 8.22 (1H, d, J = 5.4 Hz), 8.01 (1H, s), 7.88–7.92 (1H, m), 7.29–7.36 (3H, m), 6.82 (1H, d, J = 8.4 Hz), 6.88 (1H, s), 6.52 (1H, d, J = 4.9 Hz), 4.24–4.32 (2H, m), 3.01–3.04 (4H, m), 2.83–2.87 (4H, m), 2.29 (3H, s), 1.57–1.63 (3H, m). LC/MS (ESI): m/z 441 [M + H]+.
4-(1-Ethyl-3-pyridin-3-ylpyrazol-4-yl)-N-[4-(4-methylpiperazin-1-yl)phenyl]pyrimidin-2-amine (18o)The title compound 18o was prepared from compound 17 and 4-(4-methyl-piperazinyl)aniline following a procedure similar to that used to prepare compound 18b. 1H-NMR (270 MHz, CDCl3) δ: 8.85–8.86 (1H, m), 8.61 (1H, dd, J = 4.6, 1.6 Hz), 8.21 (1H, d, J = 5.1 Hz), 7.98 (1H, s), 7.91 (1H, m), 7.35 (2H, d, J = 9.2 Hz), 7.30–7.32 (1H, m), 6.85–6.89 (3H, m), 6.52 (1H, d, J = 5.4 Hz), 4.24–4.32 (2H, m), 3.16–3.19 (4H, m), 2.58–2.63 (4H, m), 2.37 (3H, s) 1.56–1.62 (3H, m). LC/MS (ESI): m/z 441 [M + H]+.
4-(1-Ethyl-3-pyridin-3-ylpyrazol-4-yl)-N-(4-morpholin-4-ylphenyl)pyrimidin-2-amine (18p)The title compound 18p was prepared from compound 17 and 4-(4-morpholinyl)aniline following a procedure similar to that used to prepare compound 18b. 1H-NMR (270 MHz, CDCl3) δ: 8.85–8.86 (1H, m), 8.61–8.62 (1H, m), 8.22 (1H, d, J = 4.9 Hz), 7.98 (1H, s), 7.86–7.91 (1H, m), 7.35–7.40 (2H, m), 7.28–7.33 (1H, m), 6.91 (1H, s), 6.83–6.87 (2H, m), 6.53 (1H, d, J = 5.1 Hz), 4.24–4.32 (2H, m), 3.85–3.89 (4H, m), 3.09–3.13 (4H, m), 1.57–1.62 (3H, m). LC/MS (ESI): m/z 428 [M + H]+
Expression and Purification of ALK2 (R206H)The ALK2 kinase domain including residues 201 to 499 with the R206H mutation was expressed with a recombinant baculovirus expression system. Sf9 cells were inoculated at 27°C and harvested 48 h after infection. The cells were resuspended in 20 mM Tris–HCl pH 8.0, 500 mM NaCl, 10% glycerol, and 20 mM imidazole and disrupted by sonication. Cell debris and insoluble components were clarified with centrifugation. The supernatant was applied to a HisTrap HP 5 mL column (GE Healthcare, Sweden) and eluted with linear gradient using 20 mM Tris–HCl pH 8.0, and 500 mM NaCl, 10% glycerol, 500 mM imidazole. The His-tag was cleaved by TEV protease and removed on the HisTrap HP 5 mL column. The buffer was exchanged to 20 mM Tris–HCl pH 8.0, 500 mM NaCl, 10% glycerol, and 2 mM dithiothreitol (DTT) for further purification. The protein was passed through a HiTrap Q-XL 5 mL column (GE Healthcare) and then purified on a 16/60 HiLoad Superdex 75 prep grade column (GE Healthcare) equilibrated with 50 mM N-(2-hydroxyethyl)piperazine-N′-2-ethanesulfonic acid (HEPES), pH 7.5, 300 mM NaCl, 50 mM L-arginine, 50 mM L-glutamate, and 10 mM DTT.
Crystallization, Data Collection and Structural AnalysisThe best crystals of ALK2 (R206H) were grown in 100 mM HEPES, pH 7.6–8.0, and 1.5–1.6 M ammonium sulfate using the sitting-drop vapor diffusion method at 20°C. Each drop contained 500 nL of protein solution at a concentration of 10 mg/mL and an equal volume of crystallization reagent. Crystals were grown spontaneously, although streak seeding helped to obtain relatively bigger crystals. The crystals were soaked in mother liquors supplemented with 5 mM compound 6a for 1 h. Then the crystals were soaked in solution containing 25% ethylene glycol and 75% of mother liquor for cryoptotection and flash-frozen under a nitrogen stream. Diffraction data were collected at the beamline BL17A of the Photon Factory (Tsukuba, Japan) at 100 K and wavelength of 0.9800 Å and processed with XDS.12) For structure determination, the software contained in the CCP4 suite13) was used. The structure of ALK2 (R206H) was determined by molecular replacement using MOLREP.14) The coordinate of the ALK2 (Q207D) mutant in complex with an inhibitor (PDBID: 3MTF) was used as the search model. Structural refinement was carried out using PHENIX,15) with iterative manual model inspection using COOT.16) Table 7 shows a summary of the results of data collection and structural refinement. The atomic coordinate and structure factor of ALK2 (R206H) in complex with compound 6a were deposited in the Protein Data Bank with the accession code 6ACR.
Table 7. Crystallographic Data and Refinement Statistics
| ALK2 (R206H)_RK-59638 (6a) |
|---|
| PDB ID | 6ACR |
| Data collection | |
| Beamline | BL-17A, Photon Factory KEK |
| Wavelength (Å) | 0.9800 |
| Space group | P21212 |
| Unit cell parameters (Å, °) | a = 85.1, b = 138.4, c = 59.4, α = β = γ = 90.0 |
| Resolution (Å)* | 50.0–2.01 (2.13–2.01) |
| Reflections* | 314633 (49976) |
| Unique reflections* | 90544 (14636) |
| Redundancy* | 3.47 (3.41) |
| Completeness (%)* | 99.6 (99.3) |
| †Rmerge* | 0.125 (0.946) |
| ‡Rr.i.m* | 0.147 (1.123) |
| I/σ(I)* | 8.59 (1.24) |
| Refinement |
| Resolution (Å) | 45.04–2.01 |
| Reflections (total) | 90502 |
| Reflections (test) | 4490 |
| Rcryst | 0.2153 |
| Rfree | 0.2568 |
| No. of atoms | |
| Protein | 4724 |
| Ligand/ion | 82 |
| Waters | 97 |
| Average B-factors (protein/ligand) (Å2) | 33.83/41.52 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.008 |
| Bond angles (°) | 0.863 |
* Values in parentheses are for the outer shell. † Rmerge = ΣhklΣi|Ii(hkl) − ⟨I(hkl)⟩|/ΣhklΣiIi(hkl). ‡ Rr.i.m = Rmeas = Σhkl(N/N − 1)1/2Σi|Ii(hkl) − ⟨I(hkl)⟩|ΣhklΣiIi(hkl).
ALK enzyme assays were conducted by Reaction Biology (United States) using the ‘HotSpot’ assay platform and kinase assay protocol. This was a 10-point assay, starting from 100 µM to 0.5 nM, performed in duplicate using the ALK inhibitor LDN-193189 as a control. In a single dose screening, the compound was tested with single dose duplicates at a concentration of 0.3 µM. The reaction was carried out at 10 µM ATP concentration. The mixture was incubated for 2 h.
P-Glycoprotein Substrate Assay Using MDR1-MDCK II CellsIn vitro efflux studies were performed at Sumika Chemical Analysis Service (Osaka, Japan), with 10 µM of compound added to either the apical (A) or basolateral (B) side of monolayers of MDCK II cells and MDCK II cells transfected with human MDR1. Apparent permeability (Papp) in the A to B and B to A directions was determined by the quantity of the compound on the opposite side of the membrane using LC/MS/MS and a fluorescence plate reader. The efflux ratio was used as an index of efflux, whereby the efflux ratio = Papp B to A/Papp A to B.
Microsomal Stability StudiesMicrosomal stability studies were performed at Sumika Chemical Analysis Service (Osaka, Japan). One micromolar of compounds with 0.5 mg/mL of liver microsomes were incubated at 37°C for 30 or 60 min in a final volume of 50 µL of 125 mM phosphate buffer solution containing 3.5 µM β-NADPH. Aliquots of incubation samples were protein precipitated with cold methanol containing labetalol (internal standard) and centrifuged, and supernatants were analyzed using LC/MS/MS. All incubations were performed in duplicate, and the percentage of parent compounds remaining at the end of incubation was determined by the LC/MS/MS peak area ratio.
PharmacokineticsThe pharmacokinetic studies were performed at Nemoto Science (Joso, Japan). The pharmacokinetics of the tested compounds were evaluated in male Sprague–Dawley rats (n = 3 or 2). The compounds were dosed intravenously at 1 mg/kg and orally by gavage at 5 mg/kg or 3 mg/kg. For the intravenous administration study, the compounds were formulated in a mixture of 10% (w/v) DMSO, 40% (w/v) PEG, and 50% (w/v) saline. For the oral administration study, the compounds were formulated as a solution in 0.5% (w/v) methyl cellulose. Blood samples were collected at eight time points (0.1, 0.25, 0.5, 1, 2, 4, 6, 24 h). Plasma proteins were precipitated with acetonitrile, and compounds concentrations were determined using LC/MS/MS. Data were analyzed using program of a moment (Excel) analysis17) and peak plasma concentration (Cmax), oral bioavailability (F), exposure (AUC), half-life (t1/2), volume of distribution (Vd), and clearance (CL) were calculated. The value of F was calculated from the ratio of the dose normalized AUC, with AUC0–t for animals dosed intravenously and AUC0–∞ for animals dosed orally.