Current Topics: Reviews
The Latest Research and Development into the Antibody–Drug Conjugate, [fam-] Trastuzumab Deruxtecan (DS-8201a), for HER2 Cancer Therapy
2019 Volume 67 Issue 3 Pages 173-185
Details
2019 Volume 67 Issue 3 Pages 173-185
A major limitation of traditional chemotherapy for cancer is dose-limiting toxicity, caused by the exposure of non-tumor cells to cytotoxic agents. Use of molecular targeted drugs, such as specific kinase inhibitors and monoclonal antibodies, is a possible solution to overcome this limitation and has achieved clinical success so far. Use of an antibody–drug conjugate (ADC) is a rational strategy for improving efficacy and reducing systemic adverse events. ADCs use antibodies selectively to deliver a potent cytotoxic agent to tumor cells, thus drastically improving the therapeutic index of chemotherapeutic agents. Lessons learned from clinical failure of early ADCs during the 1980s to 90s have recently led to improvements in ADC technology, and resulted in the approval of four novel ADCs. Nonetheless, further advances in ADC technology are still required to streamline their clinical efficacy and reduce toxicity. [fam-] Trastuzumab deruxtecan (DS-8201a) is a next-generation ADC that satisfies these requirements based on currently available evidence. DS-8201a has several innovative features; a highly potent novel payload with a high drug-to-antibody ratio, good homogeneity, a tumor-selective cleavable linker, stable linker-payload in circulation, and a short systemic half-life cytotoxic agent in vivo; the released cytotoxic payload could exert a bystander effect. With respect to its preclinical profiles, DS-8201a could provide a valuable therapy with a great potential against HER2-expressing cancers in clinical settings. In a phase I trial, DS-8201a showed acceptable safety profiles with potential therapeutic efficacy, with the wide therapeutic index.
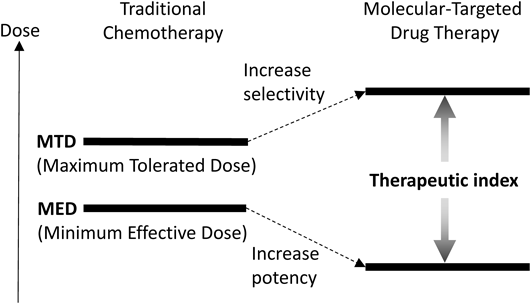
Traditional chemotherapy has been the predominant modality of cancer therapy.1) Chemotherapy agents operate by several mechanisms in which rapidly dividing cancer cells are preferentially killed, while the effect on normal cells is minimized. Historically, the next significant progress in cancer chemotherapy was the introduction of drug combinations.2,3) Suitable drug combinations with non-overlapping toxicity profiles and different mechanisms of action were found to show additive or synergistic antitumor effects. Thus, multidrug combination therapies became the standard of care for the treatment of most cancers. However, administration of therapeutic amounts of cytotoxic agents is often difficult to achieve due to their unselective and high systemic toxicity. To curtail the frequency of adverse events, next-generation approaches have been sought specifically to target molecules involved in cancer cell proliferation.4–6) These molecular targets are preferentially expressed on tumor cells relative to normal cells. Thus, molecular-targeted drugs will be selectively cytotoxic/cytostatic to tumor cells and, coupled with lower toxicity profiles, should result in a higher therapeutic indices (Fig. 1). The major approaches in targeted molecular therapy include: 1) tyrosine kinase inhibitors (TKIs); 2) monoclonal antibodies (mAbs); and 3) antibody–drug conjugates (ADCs).5–8)
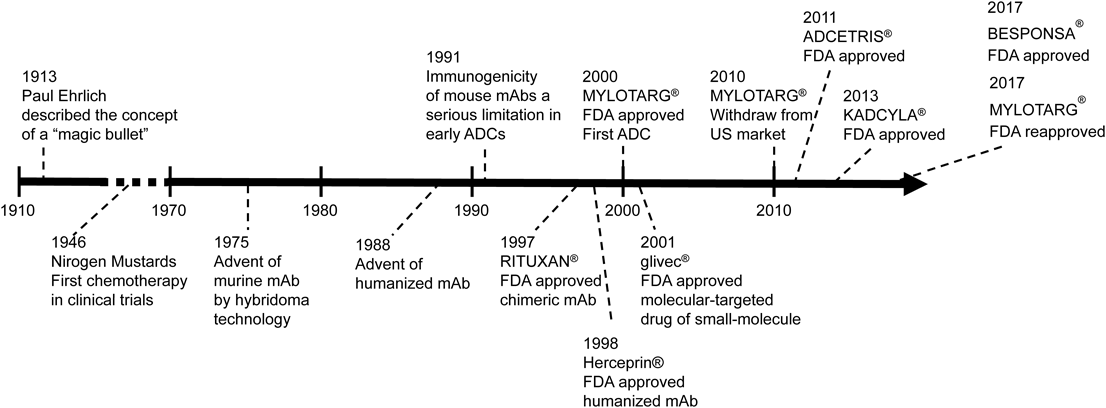
The concept of ADCs is simple. Through the connection of mAbs to small molecule drugs via an appropriate linker, the mAb, as a drug delivery carrier, and the small molecule demonstrate specific antitumor activity in cancer cells. Thus, the maximum medicinal effect in cancer cells is increased, and undesirable side effects are expected to be reduced. Historically, Paul Ehrlich proposed the concept of a “magic bullet,” as selective delivery of a cytotoxic drug to a tumor via a targeting agent a century ago.9) The history of ADC development has been challenging to the “magic bullet” imagined by Ehrlich10,11) (Fig. 2).
mAb, Monoclonal Antibody; ADC, Antibody–Drug Conjugate.
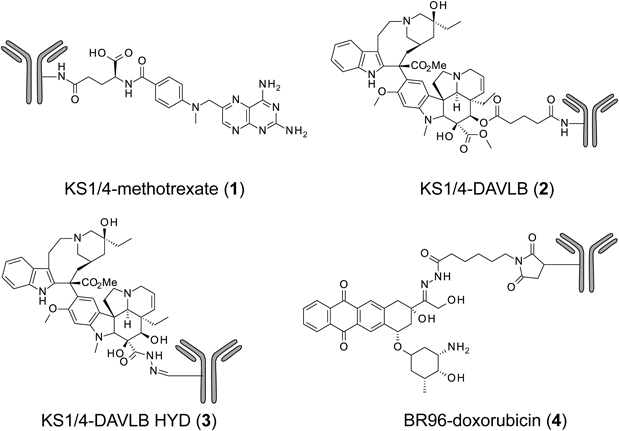
The first studies into ADCs as highly expected molecular-targeted drugs were prepared and examined clinically.7) The prepared antitumor agents to be linked to mAbs were methotrexate, vinblastine, and doxorubicin12–15) (Fig. 3). However, the early ADCs linked to these drugs did not achieve effective clinical outcomes; moreover, because the KS1/4 antibody of the ADCs [KS1/4-methotrexate (1), KS1/4-DAVLB (2), and KS1/4-DAVLB HYD (3)] was of murine origin, a majority of patients in these trials experienced an immune response to the antibodies and/or cytotoxic agents (vinca alkaloids).13,14) Although the BR96 antibody of BR96-doxorubicin (4) was a chimeric antibody, approximately 50% of the evaluable patients developed an immune response to it.15) This immune response resulted in the risk of higher clearance of the ADC from circulation during repeated dosing. However, these clinical trials provided convincing preliminary evidence for the tumor distribution of ADCs in patients. Nevertheless, the therapeutic response was insufficient. These early ADCs had low or moderate potency, compared with their potent in vitro activity, and the design of the linkers in these ADCs was not suitable as well.16)
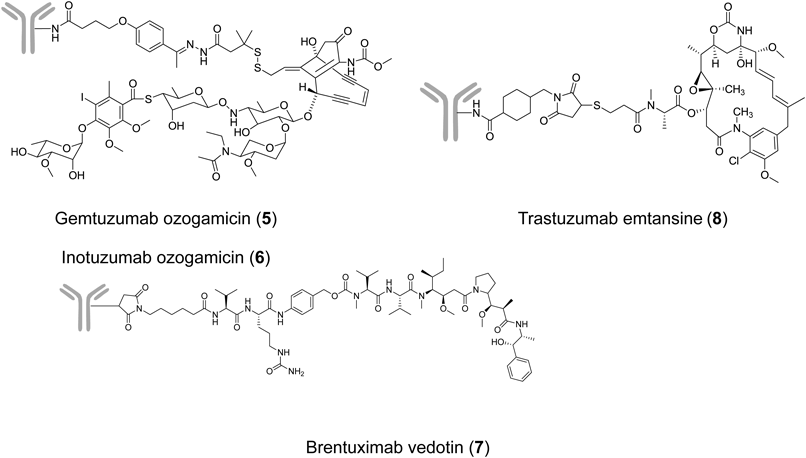
The early ADC clinical trials delivered many useful lessons-learned for ADC development7,17): more potent cytotoxic agents were required than used for existing anticancer drugs; no immune response should occur; and even greater optimization of the linker design was necessary. In 2000, the anti-acute myeloid leukemia (AML) drug gemtuzumab ozogamicin (GO; CMA-676, MYLOTARG®, 5) was the first ADC approved in the accelerated approval scheme by the Food and Drug Administration (FDA).18) GO is an anti-CD33 ADC comprising humanized immunoglobulin G4 (IgG4) antibody (hP67.6), acid-labile hybrid 4-(4′-acetylphenoxy) butanoic acid (AcBut) linker, and a calicheamicin derivative19) (Fig. 4 and Table 1). GO was indicated for the treatment of CD33-positive AML in the first relapse in patients 60 years of age or older and who were not considered candidates for cytotoxic chemotherapy. The approved dose was 9 mg/m2 intravenous (i.v.) over 4 h and repeated after 14 d. Serious adverse events of GO included fatal anaphylaxis, adult respiratory distress syndrome (ARDS), and hepatotoxicity, especially venoocclusive disease (VOD) in patients with AML.20) The drug manufacturer voluntarily withdrew the US Biologics License Application in 2010 due to the failure of the confirmatory phase III, although GO remained available in Japan.21) Recently, GO was reapproved for newly diagnosed and relapsed/refractory AML in the U.S. (2017) and Europe (2018). To reduce toxicity, investigators chose a new regimen based on repeated lower doses of GO (3 mg/m2 up to a maximum dose of 5 mg/m2) on days 1, 4, and 7 (a “3-3-3” regimen).22)
| ADC | Antibody | Linker | Drug | ||||
|---|---|---|---|---|---|---|---|
| Entry | Site DAR | Target indication | Targets antigen | Isotype | Structure trigger | Component mode of action | |
| 5 | Gemtuzumab ozogamicin | Lys 2–3 | AML | CD33 | Humanized IgG4 | AcBut hydrazone Low pH | Calicheamicin DNA-damaging agent |
| 6 | Inotuzumab ozogamicin | Lys 5–7 | ALL | CD20 | Humanized IgG4 | AcBut hydrazone Low pH | Calicheamicin DNA-damaging agent |
| 7 | Brentuximab vedotin | Cys 4 | HL ALCL | CD30 | Chimeric IgG1 | mc-VC-pABC dipeptide Lysosomal protease | Auristatin Tubulin polymerization inhibitor |
| 8 | [ado-] Trastuzumab emtansine | Lys 3.5 | Breast cancer | HER2 | Humanized IgG1 | SMCC non-cleavavle mAb degradation | Maytansine Tubulin polymerization inhibitor |
ADC: antibody–drug conjugate, DAR: drug-to-antibody ratio, IgG: immunoglobulin G, AcBut: 4-(4′-acetylphenoxy) butanoic acid, mc-VC-pABC: maleimidocaproyl valine–citrulline p-aminobenzyloxycarbamoyl, SMCC: maleimidyl 4-(N-maleimidomethyl) cyclohexane-1-carboxylate, mAb: monoclonal antibody, AML: acute myelogenous leukemia, ALL: acute lymphoid leukemia, HL: Hodgkin lymphoma, ALCL: anaplastic large cell lymphoma.
Inotuzumab ozogamicin (CMC-544, BESPONSA®, 6) is an anti-CD22 ADC composed of a humanized IgG4 antibody and the same drug-linker used for GO23) (Fig. 4 and Table 1). Inotuzumab ozogamicin was also approved by the FDA for relapsed or refractory B-cell precursor acute lymphoblastic leukemia (ALL) in 2017.24) From the clinical trial results, VOD appears a common risk from GO and inotuzumab ozogamicin. In preclinical studies, hydrazone linker-based conjugates showed stability ranges (t1/2 values) of between 2 and 3 d in mouse and human plasma, which may not be optimal for an ADC.25) Further investigation is required to elucidate the mechanism of VOD and its relation with the calicheamicin payload.26)
Brentuximab vedotin (SGN-35, ADCETRIS®, 7) is an anti-CD30 ADC conjugated with the tubulin polymerization inhibitor monomethyl auristatin E (MMAE) via a protease-cleavable dipeptide linker27,28) (Fig. 4 and Table 1). Cathepsin B is one of the enzymes responsible for linker cleavage.29) Brentuximab vedotin was approved by the FDA for the treatment of relapsed or refractory Hodgkin’s lymphoma (HL) and anaplastic large cell lymphoma (ALCL) in 2011.30,31)
[ado-] Trastuzumab emtansine (T-DM1, KADCYLA®, 8) is an anti-human epidermal growth factor receptor 2 (HER2) ADC composed of trastuzumab linked to a tubulin polymerization inhibitor maytansinoid (DM1) via the non-cleavable linker, SMCC (maleimidyl 4-(N-maleimidomethyl) cyclohexane-1-carboxylate), to a lysine residue of the antibody32,33) (Fig. 4 and Table 1). T-DM1 received FDA approval as a second-line drug for HER2-positive metastatic breast cancer (mBC) in 2013, and was the first ADC used for solid tumors.34)
Significant progress has been made in the field of ADCs for the selective delivery of cytotoxic drugs to tumors after the lessons of the early ADC failures. The early ADCs, which incorporated clinically approval drugs and mouse mAbs, had moderate activities and generally triggered immunogenic responses. On the other hand, all of four approved ADCs target overexpressed antigen, select non-immunogenic mAb carriers, apply highly potent drugs and novel stable linker technologies, and optimize drug-to-antibody ratio (DAR) in terms of efficacy and safety.
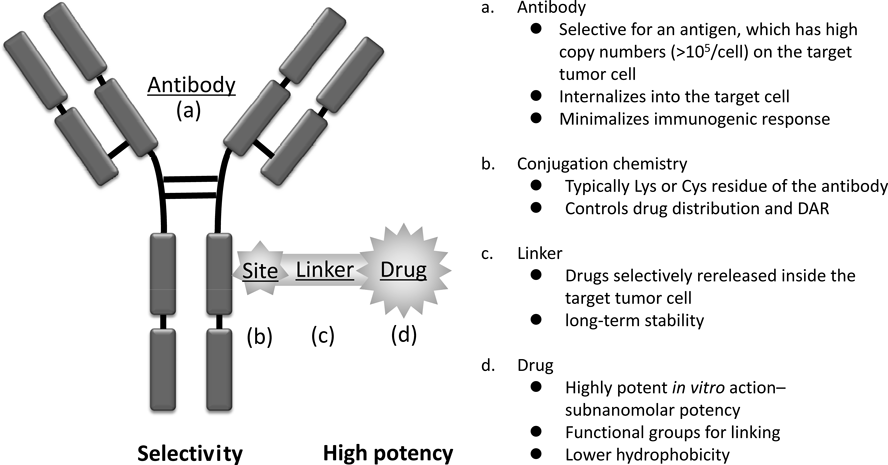
The components of ADC are a highly potent cytotoxic drug, a highly specific mAb, an appropriate linker connecting both, and a conjugation site related to drug distribution (Fig. 5). A comparative analysis of each component composed of the early and the launched ADCs is therefore expected to identify several key requirement factors.
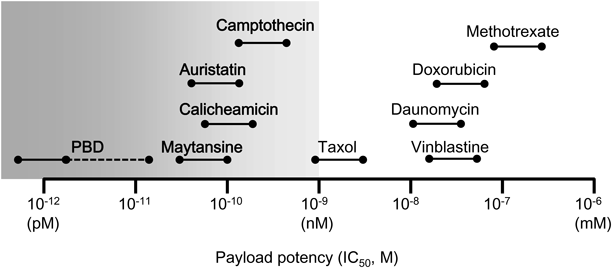
Cytotoxic agents, known as “payload,” of the early ADCs (methotrexate, vinblastine, and doxorubicin) were not sufficiently effective as ADC components.16) The delivery capacity of an antibody is dependent on two factors: the number of antigen molecules on the target cell surface and the internalization ability of ADC on binding to the antigens. The payloads used on the currently approved ADCs (calicheamicins, auristatins, and maytansinoid) are more potent than those on the early ADCs35) (Fig. 6). The required value of cytotoxic activity was generally in the subnanomolar range across a variety of human tumor cell lines. However, highly potent payloads are limited and not many have been discovered for ADC payloads. Only the DNA-damaging agent (calicheamicin) and the tubulin polymerization inhibitors (auristatins and maytansinoid) have been successfully applied in practice as the cytotoxic drugs for approved ADCs. Resistance to the approved ADCs has been reported, and there are no further treatment for tumors resistant to ADC therapies.36–39) Thus, the discovery of novel cytotoxic drugs for ADC payloads with different mechanisms of action is desirable.
PBD; Pyrrolobenzodiazepine.
Owing to heterogeneous distribution in solid tumors, antigen-negative tumor cells may escape destruction and repopulate the tumor, leading to treatment failure.40) Hence, the “bystander effect” is attracting attention.41) Bystander effect occurs when the drug from an ADC is released from the target cell following internalization and degradation of the ADC. The drug is then taken up and kills surrounding cells, which themselves may or may not express the ADC target antigen. The bystander effect depends largely on factors such as the extent of ADC internalization after binding to the target antigen and the hydrophobicity of the armed cytotoxic payload.
3.2. Linker TechnologiesThe primary emphases of linker design for ADCs are the ability to cleave active drug from the ADCs into the targeted cancer cell and the ability to remain stable in systemic circulation. The early ADCs (3–4) with an acid labile hydrazine bond were designed to release the active drug into the target cells using the acidic environment of the endosomes and lysosomes. Efficient release of cytotoxic agents was demonstrated in acidic buffer, but studies of ADC incubation under physiological conditions reported slow release of the agents.42) Because release of drug in circulation could lead to systemic toxicity and a lower therapeutic index owing to degradation of the ADCs, it was thought that an effective linker design has to balance the necessity of better stability in circulation and efficient cleavage in the target cell.
GO (5) consists of calicheamicin linked to the antibody via 4-(4′-acetylphenoxy) butanoic acid (AcBut) linker as a hydrazone bond linker.19) The AcBut linker was carefully chosen after screening a set hybrid linkers with good stability at pH 7.4 and almost complete hydrolysis at pH 4.5 at 37°C for 24 h.43)
SGN-35 (7) contains a lysosomal protease-cleavable dipeptide and self-immolative p-aminobenzyloxycarbamoyl (pABC) linker.27) The dipeptide (valine–citrulline) is recognized by cathepsins (such as cathepsin B) which are enhanced in tumors.44,45) Thus, this dipeptide was stable in circulation until it reached tumor cells and the payload was selectively released into tumor cells. Moreover, MMAE itself was released by 1,6-elimination and decarboxylation of self-immolative pABC, and exerted an antitumor effect.
In contrast, T-DM1 (8) adopted a non-cleavable linker not triggered by a drug-release mechanism to achieve greater stability in systemic circulation.33) The mechanism of drug-release is monoclonal antibody degradation, and the released cytotoxic drug from T-DM1 is lysine (Lys)-SMCC-DM1, which includes an antibody-derived Lys residue and an SMCC linker.46) Lys-SMCC-DM1 does not exert membrane permeability and has an expectation of safety while avoiding affecting normal cells. Meanwhile, Lys-SMCC-DM1 is not expected to have any bystander effect.
These approved ADCs (5–8) had permissible stability and efficacy in clinical use, but their pharmacokinetics indicated dissociation between the total antibody amount and ADC concentration.26,29,47–49) It has been shown that the linker technology of the ADC should be more stable in systemic circulation to achieve a wider therapeutic index with lower toxicity and greater efficacy.
3.3. Monoclonal AntibodyIn clinical trials of early ADCs, an immunological response was found against the antibody moiety.15) Antigenicity of antibodies in ADCs should be reduced, as with general biologics. The problem of immunogenicity can be solved with the use of “humanized” or fully human antibodies that are now readily available.50) To deliver a sufficient number of cytotoxic molecules to achieve antitumor efficacy, the antibody should be selected to target an antigen that is expressed with a high copy number (>105/cell). Additional requirements are that the antigen is not expressed or has low expression in normal cells especially in critical organs so as to avoid severe toxicity. Although a high binding affinity (KD <1 nM) may engender satisfactory localization to the tumor, it has been also suggested that an antibody with a lower binding affinity may penetrate deeply into solid tumors.51) Thus, the affinity of the antibody in ADCs should be carefully considered when screening antibodies.
The propensity for internalization into the cell to enable the intracellular delivery of the payload is also an important factor. This internalization process, called receptor-mediated endocytosis, is dependent on the nature of the antigen.52) In the case of many potential antibodies, the efficacy of each ADC in an in vivo xenograft model might yield selection of the optimal antibody despite these complicated conditions. The approach for the ADC does not require the antibody itself to possess functional activity, such as effector activity, but this function may confer additional benefits for clinical use.
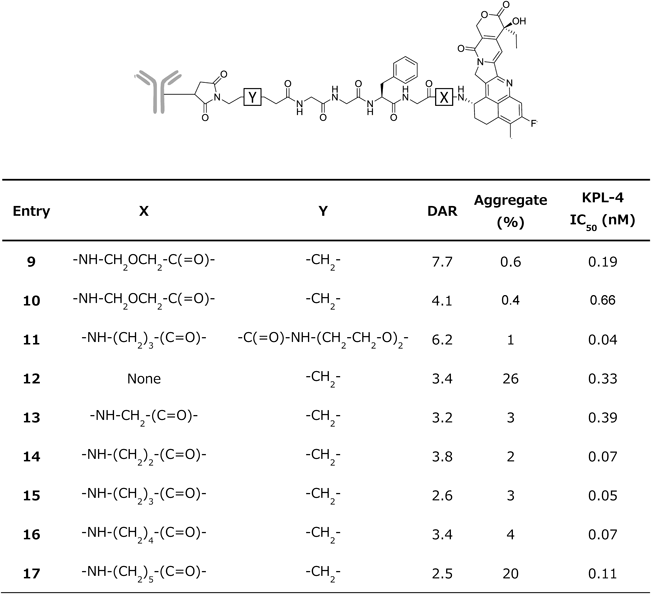
3.4. Conjugation ChemistryA typical conjugation site is a cysteine (Cys) or Lys residue present on the antibody. The primary amine of the Lys residue is a naturally nucleophilic functional group and does not require pre-functionalization prior to conjugation with the linker. It is known that approximately >20 of the >80 lysine residues on all domains of antibody are accessible as usable conjugation sites.17,53) A study found that modification of mAbs with an N-hydroxysuccinimide ester reagent yielded a population of products with DARs between 0 and 6, in which at least 40 of the lysine residues in the mAb were modified. The conjugated products potentially contained complex mixtures of over 4.5 million unique molecules.54)
In contrast, the thiol group of Cys residue is found to be a functional group with strong natural reactivity, and IgG1 has four interchain and 12 intrachain disulfide bridges.53) Because free thiols on the antibody are not present naturally, the interchain disulfide bridges often need to be selectively reduced prior to conjugation with linker, and the low abundance of only eight Cys residues on the interchain bridge allows easier control of the DAR. Since the reducing agent cannot reach intrachain disulfide bridges inside the molecule, these cannot be reduced. It was suggested that neither disruption of the interchain bridge nor the coupling of eight linkers affected clearance and stability of the antibody itself since there was no significant difference in the pharmacokinetics between unconjugated parental mAb (not reduced) and the modified mAb with Cys conjugation (interchain disulfide bonds fully reduced).55)
Although it is expected that high drug loading to an antibody may yield potent efficacy in the case of both Lys and Cys conjugation, the DAR of the approved ADCs is limited to approximately 3 to 6 owing to deterioration of the physical properties of the ADCs in systemic circulation.56) Furthermore, the optimal DAR is selected according to physicochemical properties and efficacy, however the product is usually a mixture of ADCs and these include both higher and lower DAR than the optimal number of drugs57) (Fig. 7). An approach for the synthesis of homogeneous ADCs would yield a wider therapeutic index than heterogeneous ADCs.
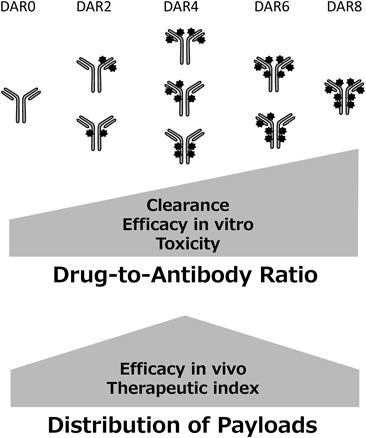
Three issues are currently topical in the ADC field. First, most of the payloads of ADC in development are limited to tubulin polymerization inhibitors and DNA-damaging agents, and there is little room for substituted therapy selection. Second, the toxicity caused by the instability of the drug-linker technology is problematic and chemical design of more stable linker technology is necessary. Third, higher drug loading is expected to lead to higher efficacy, but the number of payload is limited owing to the deterioration of physical properties. Consequently, there is an issue of heterogeneity in the drug distribution of the ADCs. Thus, improvement of existing ADC technology is necessary to achieve a wider therapeutic index, and ADC technology applicable to various antibodies is expected.
4.1. Chemistry of [fam-] Trastuzumab Deruxtecan[fam-] Trastuzumab deruxtecan (DS-8201a) is a HER2-targeting ADC under development for HER2-expressing or HER2-mutated solid tumors. DS-8201a is prepared through conjugation of a humanized anti-HER2 antibody with a topoisomerase I inhibitor payload using an enzymatically cleavable peptide-based linker58) (Fig. 8).
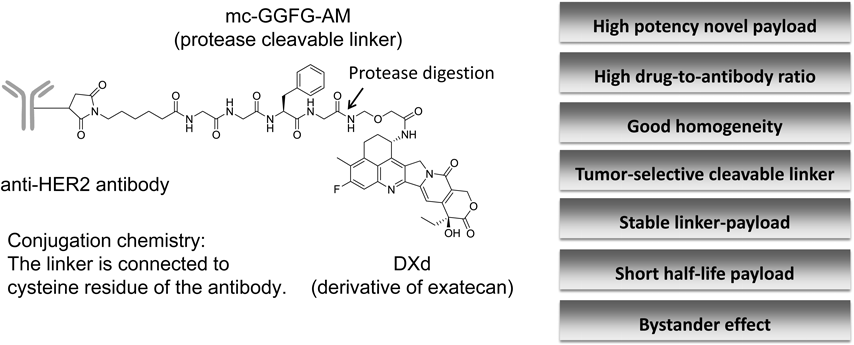
The average of DAR is 7 to 8, DXd = 2-hydroxyacetyl exatecan, mc-GGFG-AM = maleimidocaproyl glycine–glycine–phenylalanine–glycine–aminomethylene.
The antibody is a humanized IgG1 monoclonal antibody produced with reference to the amino acid sequence of trastuzumab; thus, DS-8201a binds to HER2 as well as trastuzumab. The cytotoxic payload on DS-8201a is a camptothecin (CPT) analog known as a topoisomerase I inhibitor. CPT was first isolated from the bark and stem of Camptotheca acuminata by Wall et al. in 1966.59) CPTs were able to bind and stabilize the triple-complex with DNA topoisomerase I (Topo 1) and DNA, induce DNA damage and lead to apoptosis of cells.60–62) Irinotecan and topotecan, camptothecin analogs, are used to treat several solid tumors owing to their broad-spectrum antitumor activity and cytotoxic mechanisms.63–65) Irinotecan is a prodrug that is converted into 7-ethyl-10-hydroxy-camptothecin (SN-38) by the action of carboxylesterases.65) Exatecan methanesulfonate (DX-8951f) is a water-soluble CPT that exhibits more potent topo I inhibitory activity and antitumor activity than other CPT analogs. Furthermore, exatecan is effective against P-glycoprotein (P-gp)-mediated multi-drug resistant cells.66) Various linkers and exatecan derivatives, which are highly efficient CPT analogs, were investigated67) (Table 2). The topoisomerase I inhibitory potency of the novel exatecan derivative (DXd, 19) was 10-fold higher than SN-38.58) DXd is therefore expected to have sufficient efficacy when used as a payload for ADCs.
 |
DS-8201a achieved a higher DAR of approximately 8 with homogeneous conjugation compared with all currently approved ADCs57,58,68,69) (Fig. 9). The DAR of 8 is theoretically the maximum drug loading number for conventional interchain Cys conjugation. Despite the high DAR, the pharmacokinetic profile of DS-8201a in cynomolgus monkeys was favorable58) (Fig. 10A). When DS-8201a was intravenously administered at 3 mg/kg, there was no clear difference between DS-8201a and the total antibody and DXd was detected only at limited time points and at much lower concentrations than DS-8201a, indicating that the linker-payload of DS-8201a is stable in plasma. An in vitro study of stability in plasma was performed in several species. The release rate of DXd from DS-8201a in human plasma was as low as 2.1% after 21 d of incubation58) (Fig. 10B), whereas the release rate of T-DM1, an anti-HER2 ADC, was 18.4% after incubation for 4 d.70)
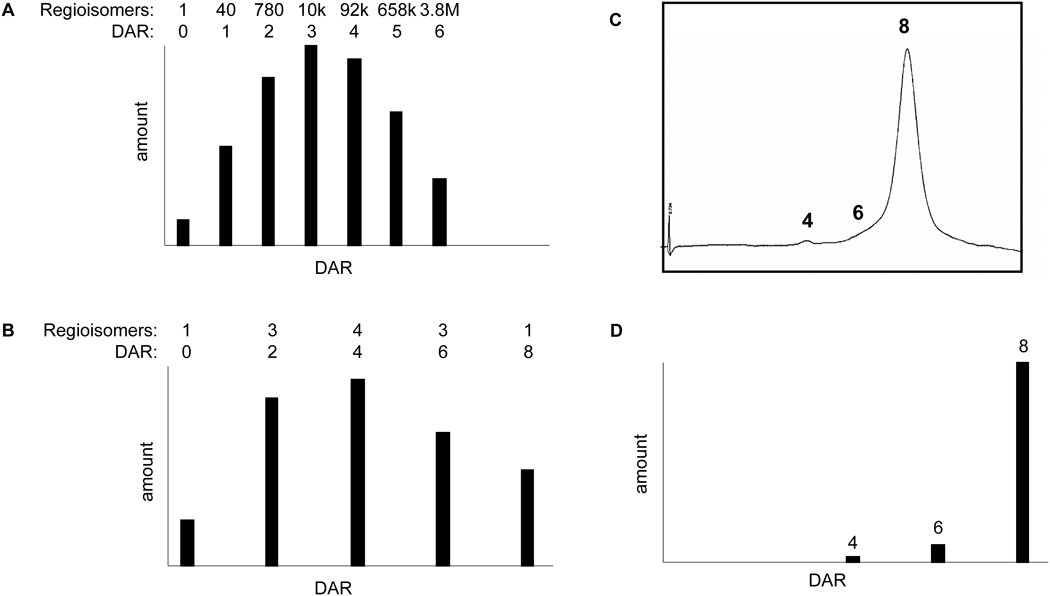
A: Distribution of drug-linkers on lysine conjugates. B: Distribution of drug-linkers on cysteine conjugates. C: Hydrophobic interaction chromatography of DS-8201a. D: Distribution of conjugated drugs of DS-8201a. DAR: Drug-to-antibody ratio.
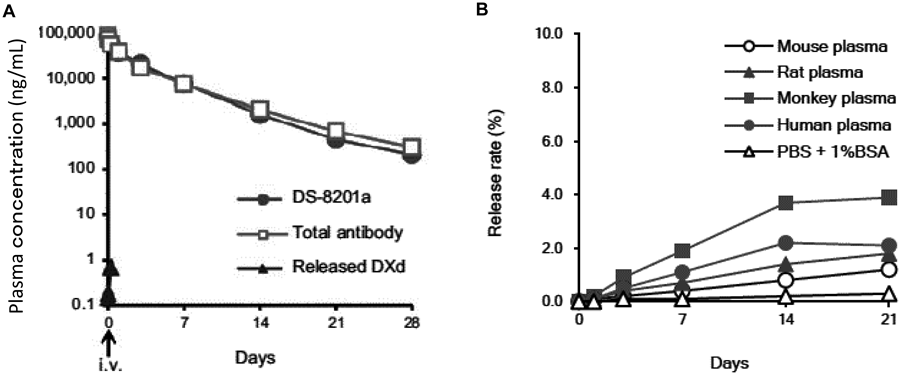
A: Pharmacokinetics of DS-8201a in cynomolgus monkeys. Mean plasma concentrations of DS-8201a, total antibody, and DXd after single intravenous administration of DS-8201a at 3 mg/kg to cynomolgus monkeys (n = 3). B: In vitro plasma stability of DS-8201a. PBS: Phosphate buffered saline, BSA: Bovine serum albumin.
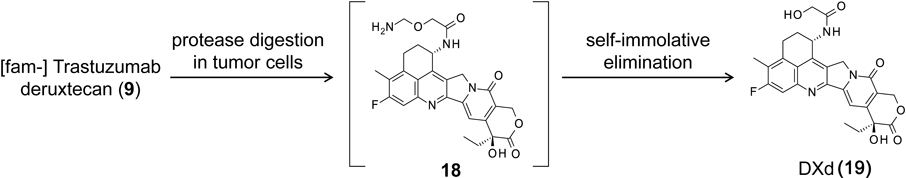
The linker between the antibody and payload is an enzymatically cleavable peptide (GGFG)-based linker,71) and a self-immolative amino methylene spacer, which reduced hydrophobicity compared with the conventional p-amino benzyl (pAB) spacer and provided stability in systemic circulation. The mechanism of drug release is shown in Fig. 11. First, the internalized ADC is selectively cleaved to a temporary hydrolysate (18) composed of DXd by lysosomal proteases in tumor cells. Subsequently, an amino-methylene (AM), which is designed as a self-immolative spacer, is rapidly hydrolyzed to ammonia and formaldehyde. Finally, intracellular release of DXd (19) occurs to trigger cell death. Moreover, DXd was designed to have good cell membrane permeability so as to show a bystander effect of the ADC.72)
The seven structural features of DS-8201a are: a high potency novel payload; high DAR; good homogeneity; tumor-selective cleavable peptide linker; stable linker-payload in circulation; short half-life of the cytotoxic agent in vivo; and bystander effect of the released cytotoxic payload.
4.2. Preclinical Studies of [fam-] Trastuzumab DeruxtecanHER2 is a member of the epidermal growth factor receptor (EGFR) family of transmembrane receptors and overexpressed in a broad number of cancer types, such as breast, colorectal, gastric, lung, and ovarian cancers.73–75) Overexpression of HER2 [immunohistochemistry (IHC) 3+ or IHC 2+/in-situ hybridization (ISH)+] was observed in 10–20% of breast cancer cases.76) In addition, low levels of HER2 (IHC2+/ISH− or IHC1+) were reported approximately 50% in breast cancer.77) T-DM1 is known to show antitumor efficacy against HER2-positive breast cancers, but not against tumors with low HER2 expression.77)
The efficacy of DS-8201a was compared with T-DM1 and a lower-DAR HER2 ADC (DAR 3.4, the same the DS-8201a linker-payload) in mice xenograft models with different levels of HER2 expression58) (Fig. 12). T-DM1 showed efficacy only in KPL-4, a breast cancer cell line with high HER2 expression. In contrast, DS-8201a was effective in the cell line Capan-1, a pancreatic cell line with low HER2 expression, in addition to efficacy in KPL-4 and JIMT-1 (a T-DM1 refractory HER2-positive breast cancer cell line). Neither ADCs were effective in the GCIY cell line, which is a gastric cancer cell line negative for HER2 expression. An ADC of DAR 3.4 indicated antitumor activity against all levels of HER2 expression, but the degree was dependent on the expression of HER2, and the activity of an ADC of DAR 3.4 in low-expressing HER2 Capan-1 cells was weaker than that of DS-8201a with a DAR of 8. These results showed that the high DAR of DS-8201a can deliver more drug into tumor cells compared with T-DM1 and a DAR of 3.4, and DS-8201a showed preliminary evidence of activity in low-expressing HER2 tumors.
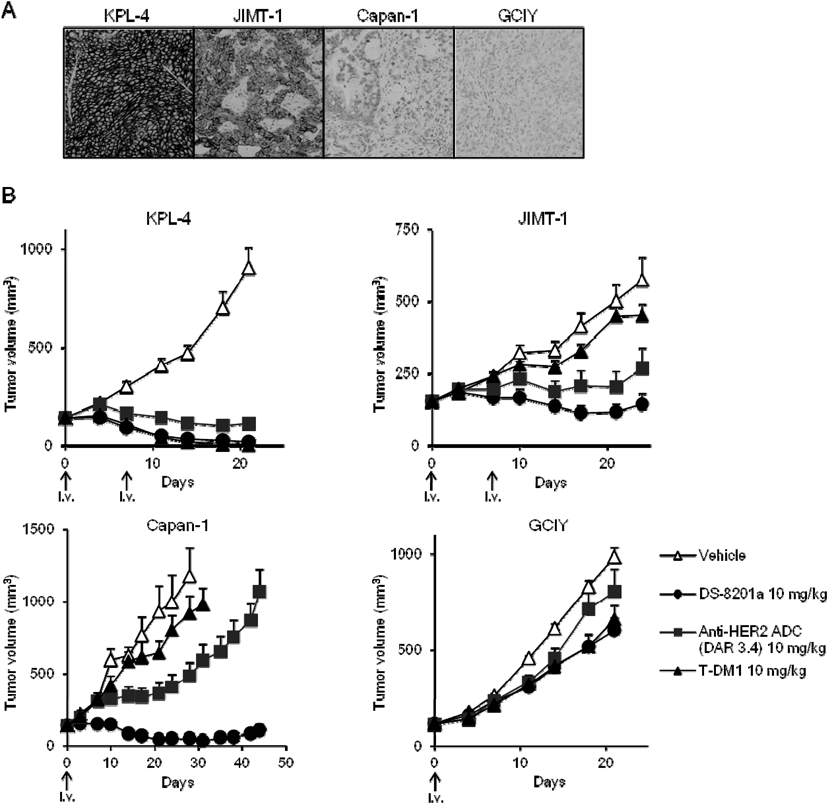
The released DXd exerts antitumor activity through cytotoxic activity not only in the target cells, but also in neighboring tumor cells owing to the membrane permeability of the released cytotoxic payload72) (Fig. 13). Thus, DS-8201a is expected to show a high therapeutic efficacy in heterogeneous tumors owing to the bystander effect. DXd is also expected to have an acceptable safety profile based on its short half-life in systemic circulation as observed in pre-clinical study58) (Fig. 10A).
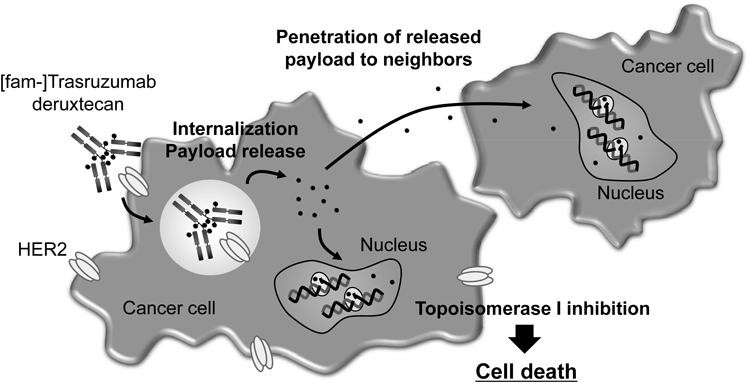
From these results, DS-8201a may be effective even in tumors with low HER2 expression levels because it can efficiently deliver the cytotoxic payload owing to a high DAR and high linker stability and be effective in a heterogeneous tumor environment owing to the bystander effect. The potent antitumor efficacy of DS-8201a has been confirmed in several patient-derived xenograft models, including breast and gastric cancers.58)
DS-8201a was well tolerated in rats as the non-cross reactive species and in monkeys as the cross reactive species58) (Table 3). When DS-8201a was administered at doses 20–197 mg/kg for rats and 10–78.8 mg/kg for monkeys every 3 weeks for a total of three doses, the severely toxic dose in 10% of the animals (STD10) was >197 mg/kg in rats and the highest non-severely toxic dose (HNSTD) was 30 mg/kg in monkeys.
| Species | Crl: CD(SD) rats | Cynomolgus monkeys |
|---|---|---|
| Dosage | 0, 20, 60, and 197 mg/kg | 0, 10, 30, and 78.8 mg/kg |
| Regimens | Intravenous, every 3 weeks | Intravenous, every 3 weeks |
| Days 1, 22, 43 (total three doses) | Days 1, 22, 43 (total three doses) | |
| No. of animals | 10/sex/group (Main): all doses | 3/sex/group (Main): all doses |
| 5/sex/group (Recovery): 60 and 197 mg/kg | 2/sex/group (Recovery): 30 and 78.8 mg/kg | |
| Lethal dose | >197 mg/kg | 78.8 mg/kg (1 female died) |
| Body weight | 197 mg/kg: low body weight gain | 78.8 mg/kg: decreased in 1 male and 1 female |
| Hematology | 60 mg/kg: decreased RBC and WBC parameters | 78.8 mg/kg: decreased RBC parameters |
| Target organs and tissues | 20 mg/kg: intestines, testes | 10 mg/kg: intestines |
| 60 mg/kg: bone marrow, thymus, lymph nodes, skin, kidneys, incisors | 30 mg/kg: lungs, skin, testes | |
| 78.8 mg/kg: bone marrow, kidney | ||
| STD10/HNSTD | STD10: >197 mg/kg | HNSTD: 30 mg/kg |
RBC, red blood cell; WBC, white blood cell; STD10, severely toxic dose in 10% of animals; HNSTD, highest non-severely toxic dose.
The results of these preclinical studies have provided strong evidence to support the potent antitumor activity and favorable pharmacokinetic and safety profiles of DS-8201a in clinical studies.
4.3. Clinical Study of [fam-] Trastuzumab DeruxtecanOverall, 10–20% of breast cancer tumors are HER2-positive, with or without hormone receptor expression.76) These patients are classified as having HER2-positive breast cancers, which tend to grow and spread more aggressively. Therefore, a number of drugs have been developed to target this protein. Trastuzumab (Herceptin®) is the humanized anti-HER2 IgG1 monoclonal antibody.79) This epoch-making drug substantially improved patient survival. However, despite treatment with trastuzumab and common chemotherapy drugs, patients continued to show signs of disease progression, whereas T-DM1 significantly prolonged PFS and overall survival (OS) with lower toxicity than lapatinib plus capecitabine in patients with HER2-positive advanced breast cancer previously treated with trastuzumab and a taxane.34) Thus, T-DM1 continues to benefit many patients as a second-line treatment option for advanced HER2-positive mBC.
In the dose-escalation part of DS-8201a phase I trial,80) 24 patients received initial intravenous doses of DS-8201a between 0.8 and 8.0 mg/kg (n = 3 for the 0.8, 1.6, 3.2, and 8.0 mg/kg doses; n = 6 for 5.4 and 6.4 mg/kg doses), with dose-limiting toxicity (DLT) assessed on a 21-d cycle. DLT was not observed and the maximum tolerated dose (MTD) was not reached, even at 8.0 mg/kg, which is the highest dose of trastuzumab used clinically. The most common adverse events were mild or moderate gastrointestinal and hematological events.
Analysis of the pharmacokinetic properties showed a non-linear pharmacokinetic profile and a longer half-life at higher doses. Serum concentrations of intact DS-8201a, free DXd, and total antibody versus time after treatment with 6.4 mg/kg DS-8201a are shown in Fig. 14.80) Low concentrations of free DXd were observed and the total antibody concentrations similar to DS-8201a concentrations at all time points assessed, suggesting a high stability of the linker-payload in systemic circulation, as similar to the pre-clinical result.
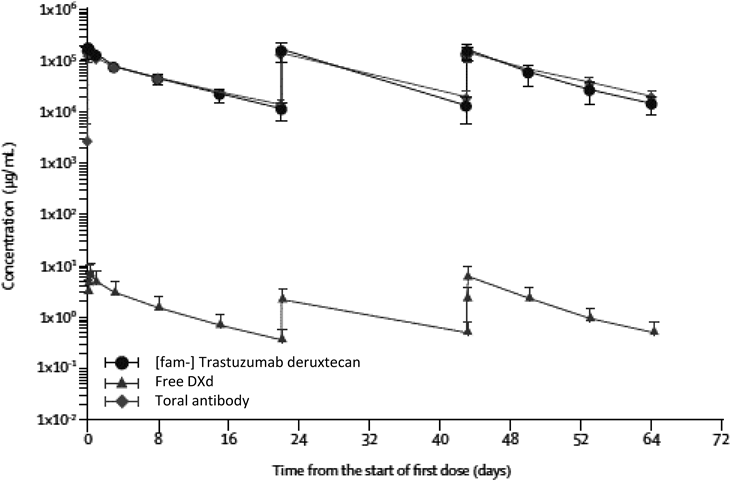
Mean serum concentration versus time curve of [fam-] trastuzumab deruxtecan, free DXd, and total antibody over cycles 1–3 at a dose of 6.4 mg/kg. Error bars indicate S.D.
On the basis of the balance between efficacy and safety, the 5.4 and 6.4 mg/kg doses of DS-8201a monotherapy were selected for further investigation into the dose expansion part. In the dose expansion part of the Phase I study,81) DS-8201a was administered to patients with T-DM1-treated HER2-positive breast cancer, trastuzumab-treated HER2-positive gastric cancer, low-expressing HER2 breast cancer, or HER2-expressing or HER2-mutated solid tumors.
Based on an interim analysis of Phase I, with a data cutoff of April 18, 2018 (Table 4), an updated subgroup analysis of 99 patients with HER2-positive mBC pretreated with trastuzumab and T-DM1 (as well as pertuzumab in the majority of cases) demonstrated a 54.5% (54/99 patients) confirmed overall response rate (ORR) and a 93.9% (93/99 patients) disease control rate (DCR). Furthermore, an updated subgroup analysis in 34 heavily pretreated patients with HER2-low-expressing mBC, defined as IHC 2+/ISH- and IHC 1+, demonstrated a 50.0% (17/34 patients) confirmed ORR and an 85.3% (29/34 patients) DCR treated with DS-8201a. In the subgroup of 44 patients with HER2-positive gastric cancer or gastroesophageal junction adenocarcinoma previously treated with trastuzumab and chemotherapy, a confirmed ORR of 43.2% (19/44 patients) and a DCR of 79.5% (35/44 patients) were observed after DS-8201a treatment.
| HER2-Pos. BC N = 99 | HER2-Low BC N = 34 | HER2-Pos. GC N = 44 | Other cancers N = 31 | |
|---|---|---|---|---|
| Confirmed ORR, % (n/N) | 54.5% (54/99) | 50.0% (17/34) | 43.2% (19/44) | 38.7% (12/31) |
| DCR, % (n/N) | 93.9% (93/99) | 85.3% (29/34) | 79.5% (35/44) | 83.9% (26/31) |
| Median PFS, months (95% CI) | NR | 12.9 (NA) | 5.6 (3.0, 8.3) | 12.1 (2.7, 14.1) |
| min, max (months) | 1.0, 22.2+ | 0.5, 19.6+ | 1.2, 19.6+ | 0.7, 14.1+ |
Dose: 5.4 or 6.4 mg/kg, BC, breast cancer; GC, gastric/gastroesophageal junction cancer; CI, confidence interval; DCR, disease control rate; HER2, human epidermal growth factor receptor 2; Pos., Positive; NA, not available; NR, not reached; ORR, overall response rate; PFS, progression-free survival. The data cutoff for this analysis was April 18, 2018.
In addition, confirmed responses were observed across a range of other HER2-expressing or HER2-mutant solid tumors, including NSCLC, CRC, and salivary gland carcinomas. In all 241 patients, the most common adverse events were gastrointestinal or hematologic in nature (Table 5) and the safety data suggested that, in general, DS-8201a has an acceptable safety profile.
| All grades | Grade ≥3 | |
|---|---|---|
| Nausea | 166 (68.9) | 6 (2.5) |
| Vomiting | 84 (34.9) | 4 (1.7) |
| Diarrhea | 64 (26.6) | 2 (0.8) |
| Constipation | 51 (21.2) | 0 (0.0) |
| Stomatitis | 43 (17.8) | 0 (0.0) |
| Decreased appetite | 134 (55.6) | 8 (3.3) |
| Anemia | 77 (32.0) | 36 (14.9) |
| Decreased platelet count | 69 (28.6) | 25 (10.4) |
| Decreased neutrophil count | 61 (25.3) | 37 (15.4) |
| Decreased white blood cell count | 58 (24.1) | 30 (12.4) |
| Alopecia | 87 (36.1) | 0 (0.0) |
| Fatigue | 67 (27.8) | 4 (1.7) |
| Malaise | 50 (20.7) | 1 (0.4) |
| Pyrexia | 25 (10.4) | 1 (0.4) |
| Dysgeusia | 24 (10.0) | 0 (0.0) |
Dose: 5.4 or 6.4 mg/kg, N = 241. The data cutoff for this analysis is April 18, 2018.
DS-8201a is currently in pivotal phase II clinical development for HER2-positive unresectable and/or metastatic BC resistant or refractory to T-DM1 (DESTINY-Breast01, NCT03248492) in North America, Europe, and Asia, and in pivotal phase II development for HER2-positive advanced gastric cancer resistant or refractory to trastuzumab (DESTINY-Gastric01, NCT03329690) in Japan and South Korea. In addition to these pivotal phase II studies, global phase II studies in patients with advanced HER2-expressing colorectal cancer (NCT03384940), and advanced HER2-overexpressing or HER2-mutated non-small cell lung cancer are ongoing (NCT03505710), as well as a Phase Ib combination study with the anti-PD1 monoclonal antibody nivolumab (NCT03523572).
Initially, the simple drug delivery concept in which ADCs selectively directed a potent drug to tumors attracted tremendous attention, and many research and development activities led to clinical trials in the 1980s and 1990s. However, clinical trials of the early ADCs resulted in failure to meet these great expectations. The root cause analysis and improvements on previous clinical failures led to four approved ADCs. The main improvements are that antibodies are human or humanized not immunogenic, and linkers are designed to be not only more stable in systemic circulation, but also cleaved in tumor cells alone. Moreover, potent cytotoxic agents that could not be used with small-molecule chemotherapy could be adapted for ADC payloads owing to targeted drug delivery. Recently, more than 70 different ADCs have been investigated in clinical development and different drug-linker systems from the approved ADCs have been applied to some ADCs. The next-generation ADCs will have further improvements, such as novel cytotoxic drugs other than tubulin polymerization inhibitors and DNA-damaging agents, more stable linker technology in systemic circulation, a higher loading number of drugs, or site-specific conjugation for homogeneity.
[fam-] Trastuzumab deruxtecan (DS-8201a) is a next-generation ADC that attempts to solve previous challenges. DS-8201a is a HER2-targeting ADC structurally composed of a humanized anti-HER2 antibody, an enzymatically cleavable peptide-based linker, and a novel topoisomerase I inhibitor (DXd). The anti-HER2 antibody component of DS-8201a is a human monoclonal IgG1 produced with reference to the same amino acid sequence as trastuzumab. A self-immolative amino methylene spacer was used in DS-8201a to reduce hydrophobicity and provide stability in systemic circulation. Therefore, this ADC achieves a high drug-to-antibody-ratio (DAR approximately 8) with good homogeneous conjugation to DXd. DS-8201a differs from T-DM1 in several ways: 1) different mechanism of action, 2) antitumor efficacy in HER2 low expressing tumors owing to high DAR and stability, and 3) antitumor activity in heterogenic tumors owing to the bystander effect. The Phase II/III DESTINY studies of DS-8201a are currently underway. The translation from the preclinical and phase I trials of this new topoisomerase I inhibitor-based ADC technology into the clinical proof-of-concept will be clarified in the studies. In addition, because the preclinical data indicate a possibility for the combination of topoisomerase I inhibitor-based ADC technology with immuno-oncology therapy for better outcomes,82) this clinical combination is currently under investigation. DS-8201a might provide valuable therapy with the potential to respond to T-DM1-insensitive breast cancer and other HER2 expressing cancers in clinical practice.
We thank the patients who participated in DS-8201 study, as well as their families and caregivers. We gratefully acknowledge the work of past and present co-workers of the DS-8201 project team.
The authors are employees of Daiichi Sankyo Co., Ltd.