Abstract
In order to develop an efficient organocatalyst for the enantioselective N–H insertion reaction via carbene/carbenoid, the catalytic core of the cinchona alkaloids was investigated. According to our working hypothesis of an eight-membered ring transition state in the N–H insertion reaction, two pairs of enantiomers related to 2-amino-1-phenylethanol were investigated for their chiral inducing potential. Since both (1R,2S)-isomers gave the N-phenyl-1-phenylglycine derivative enriched in the R-form, while their enantiomers gave the S-form, the 2-amino-1-phenylethanol structure is concluded to be the catalytic core of the cinchona alkaloid in the enantioselective N–H insertion reaction via rhodium(II) carbenoid.
Introduction
The stereoselective N–H insertion reaction is one of the most attractive subjects in medicinal chemistry because pharmacological events of amino compounds depend on chirality.1–3) While highly stereoselective C–H and Si–H insertion reactions using α-diazocarbonyl compounds have been achieved by well-defined rhodium(II) carboxylates4–6) and carboxamides7) as chiral catalysts, there are a few examples of enantioselective N–H insertion reactions initiated by rhodium(II) complexes using either chiral catalysis8,9) or cooperative catalysis.10–13) In cooperative catalysis for X–H (X = N, O, etc.) insertion reactions, cinchona alkaloids or chiral phosphoric acids are used in combination with achiral rhodium(II) carboxylates such as dirhodium(II) tetrakis(triphenylacetate), Rh2(TPA)4,10,11,13) or dirhodium(II) tetrakis(trifluoroacetate), Rh2(TFA)4.12) Cinchona alkaloids have been used widely in many types of organic transformations to generate products with high optical purities.14,15) In addition to the naturally occurring cinchona alkaloids, semi-synthetic analogs have exhibited excellent utility in organic chemistry.16)
Results and Discussion
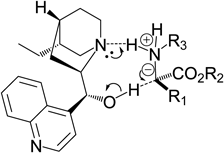
We previously reported the enantioselective N–H insertion reaction on aniline with either a carbene or carbenoid in the presence of a catalytic amount of a cinchona alkaloid.10,17) The highly reactive carbene or carbenoid was generated from phenyldiazoacetate by heating17) or treating with a dirhodium(II) complex,10) respectively. Since the enriched (R)-phenylglycine derivative was produced by hydrocinchonine, whereas its pseudo-isomer, hydrocinchonidine, predominantly gave the (S)-form, we concluded that the chiral induction in these N–H insertion reactions was solely dependent on the chirality of the cinchona alkaloid. This chiral induction suggests that the carbene/carbenoid forms an ammonium ylide with aniline, which then undergoes a [1,2]-proton shift through an eight-membered ring transition state, as shown in Fig. 1. In order to optimize an efficient artificial organocatalyst, we planned to clarify the catalytic core of the cinchona alkaloid in the carbene-driven N–H insertion reaction. According to the structure of the transition state, the tert-amine and sec-alcohol moieties in the vicinal carbons are coordinated with ammonium and the anionic carbon in the ylide-type intermediate, respectively. The aromatic ring and quinuclidine moiety may cause the conformation of the catalyst to be rigid by steric hindrance and restrict the orientation of both the amine and the alcohol. In this paper, we describe the potential of the chiral 2-aminoethanol, which is functional groups included in the structure of hydrocinchonine (Fig. 2).
To investigate the minimum requirement of the cinchona alkaloid catalysts for the enantioselective N–H insertion reaction, we chose two pairs of chiral 2-amino-1-phenylethanol derivatives. One pair was (1R,2S)- and (1S,2R)-2-(dibutylamino)-1-phenylpropan-1-ol (1a, 1b) and the other was (1R,2S)- and (1S,2R)-1-phenyl-2-(pyrrolidin-1-yl)propan-1-ol (2a, 2b)18,19) (Fig. 3). Fortunately, when the rhodium carbenoid generated from methyl phenyldiazoacetate (3a) and dirhodium(II) tetrakis(triphenylacetate) [Rh2(TPA)4, 1 mol%] was subjected to the N–H insertion reaction with aniline (4a) in the presence of a (1R,2S)-aminoalcohol, methyl 2-phenyl-2-(phenylamino)acetate (5a) was obtained enriched in the R-isomer (Table 1). Methylene chloride was found to be the most suitable solvent among the four solvents tested, and the highest chemical yields (99%) with enantioselectivities (51%) were observed when 2 mol% of 1a and 1.2 eq of aniline were used (Entry 7). Interestingly, when the reaction was conducted at 0°C, the enantioselectivity decreased to 39% enantiomeric excess (ee), as did the chemical yield (Entries 7 vs. 9). When the reaction temperature was increased to the boiling point of methylene chloride, the chemical yield and enantioselectivity decreased slightly (Entries 7 vs. 10).
Table 1. Enantioselective N–H Insertion Reaction of Phenyldiazoacetate and Aniline Using Rh(II) and Chiral 2-Amino-1-phenylethanol Derivatives
a) |
|---|
| Entry | Rh(Il) | Organocatalyst (mol%) | Aniline (equiv.) | Solvent | α-Amino esters |
|---|
| Yield, %b) | Ee, %c) |
|---|
| 1 | Rh2(TPA)4 | Hydrocinchonine (1) | 1.2 | CH2Cl2 | 83 | 58 (R) |
| 2 | Rh2(TPA)4 | la (10) | 1.2 | CH2Cl2 | 96 | 48 (R) |
| 3 | Rh2(TPA)4 | la (10) | 1.2 | CHC13 | 87 | 33 (R) |
| 4 | Rh2(TPA)4 | la (10) | 1.2 | Toluene | 91 | 44 (R) |
| 5 | Rh2(TPA)4 | la (10) | 1.2 | Hexane | 45 | 21 (R) |
| 6 | Rh2(TPA)4 | la (5) | 1.2 | CH2Cl2 | 97 | 50 (R) |
| 7 | Rh2(TPA)4 | la (2) | 1.2 | CH2Cl2 | 99 | 51 (R) |
| 8 | Rh2(TPA)4 | la (1) | 1.2 | CH2Cl2 | 95 | 45 (R) |
| 9d) | Rh2(TPA)4 | la (2) | 1.2 | CH2Cl2 | 75 | 39 (R) |
| 10e) | Rh2(TPA)4 | la (2) | 1.2 | CH2Cl2 | 92 | 45 (R) |
| 11 | Rh2(TPA)4 | la (2) | 2.0 | CH2Cl2 | 88 | 45 (R) |
| 12 | Rh2(TPA)4 | la (2) | 5.0 | CH2Cl2 | 74 | 38 (R) |
| 13 | Rh2(S-PTTL)4 | la (2) | 1.2 | CH2Cl2 | 86 | 43 (R) |
| 14 | Rh2(S-PTTL)4 | — | 1.2 | CH2Cl2 | 89 | rac |
| 15 | Rh2(R-PTTL)4 | la (2) | 1.2 | CH2Cl2 | 89 | 46 (R) |
| 16 | Rh2(TPA)4 | lb (2) | 1.2 | CH2Cl2 | 96 | 51 (S) |
| 17 | Rh2(TPA)4 | 2a (2) | 1.2 | CH2Cl2 | 93 | 31 (R) |
| 18 | Rh2(TPA)4 | 2b (2) | 1.2 | CH2Cl2 | 88 | 33 (S) |
a) All the reactions were carried out as follows; A mixture of diazocompound 3 (0.2 mmol) and aniline in the indicated solvent (2 mL) was added to a solution of Rh(II) complex and organocatalyst in the same solvent (2 mL). b) Isolated yield. c) Determined by HPLC (Daicel Chiralpak AD-H). d) The reaction was carried out at 0°C. e) The reaction was carried out at reflux.
Next, we addressed whether the chirality of the rhodium complex used for generating the carbenoid influenced the chirality of the product. For this purpose, a pair of chiral dirhodium(II) tetracarboxylates [Rh2(PTTL)4]20–24) was used for the carbenoid generation. In the N–H insertion reaction using either Rh2(S-PTTL)4 or Rh2(R-PTTL)4 in the presence of aminoalcohol (1a), the R-form of the product was generated in 43% and 46% ee, respectively (Entries 13 and 15), while the N–H insertion reaction in the absence of aminoalcohol (2a) gave a racemic mixture (Entry 14). These results suggested that the chirality of the product is only due to the chirality of the organocatalyst and that the rhodium complex does not influence the formation of the eight-membered ring transition state17) (Fig. 4). We compared the potential of four aminoalcohols (1a, 1b, 2a, 2b) as organocatalysts in the enantioselective N–H insertion reaction. The chemical yields were nearly quantitative (88–99%) in all cases. In these reactions, (1R,2S)-conformers (1a, 2a) gave the enriched R-enantiomer of 5a, 51% ee with 1a and 31% ee with 2a (Entries 7 vs. 17), while (1S,2R)-conformers (1b, 2b) gave the enriched S-enantiomer of 5a, 51% ee with 1b and 33% ee with 2b (Entries 16 vs. 18).
We then investigated the substrate scope of the reaction with respect to the ester groups and aniline component (Table 2). In all cases, high yield and similar degree of enantioselectivities were consistently observed. However, no reaction occurred when p-anisidine was used (Entry 9).
Table 2. Enantioselective N–H Insertion Reaction of Phenyldiazoacetate and Aniline Using Rh(II) and Chiral 2-Amino-1-phenylethanol Derivatives
a) |
|---|
| Entry | Diazo | Aniline | α-Amino esters |
|---|
| R1 | | R2 | | Yield, %a) | Ee, %b) |
|---|
| 1 | 3a | Me | 4a | H | 5a | 99 | 51 |
| 2 | 3b | Et | 4a | H | 5b | 94 | 43c) |
| 3 | 3c | isoBu | 4a | H | 5c | 90 | 42 |
| 4 | 3a | Me | 4b | F | 5d | 90 | 40 |
| 5 | 3a | Me | 4c | Cl | 5e | 94 | 45 |
| 6 | 3a | Me | 4d | Br | 5f | 94 | 45 |
| 7 | 3a | Me | 4e | I | 5g | 93 | 41 |
| 8 | 3a | Me | 4f | Me | 5h | 95 | 40 |
| 9 | 3a | Me | 4g | OMe | | No reaction | |
a) Isolated yield. b) Determined by HPLC (Daicel Chiralpak AD-H) unless otherwise stated. c) Determined by HPLC (Daicel Chiralcel OJ-H).
We performed additional study to obtain information on the reaction mechanism. We prepared O-methyl analog (1c) of β-aminoethanol catalyst.25) The N–H insertion product 5a was obtained in 94% yield as a racemic compound (Chart 1). The loss of enantioselectivity suggests the participation of the secondary hydroxyl group for the enantiomeric induction. This result supports the proposed transition state as shown in Fig. 4.

Chart 1
In summary, we have demonstrated a catalytic system of Rh2(TPA)4 and simple chiral aminoethanol is effective for enantiomeric induction in intermolecular N–H insertion reactions. We are continuing to pursue more efficient catalytic systems in terms of enantioselectivities.
Acknowledgments
This work was in part supported by the “Private University Research Branding Project” from the Ministry of Education, Culture, Sports, Science and Technology (MEXT). We thank Dr. Koichi Metori (Chemical Analysis Center, School of Pharmacy, Nihon University) for performing mass measurements. We also thank Mr. Yudai Tanaka, Mr. Yukio Fukuzono, and Mr. Syunta Tayama for technical assistance.
Conflict of Interest
The authors declare no conflict of interest.
Supplementary Materials
The online version of this article contains supplementary materials.
References
- 1) Moody C. J., Angew. Chem. Int. Ed., 46, 9148–9150 (2007).
- 2) Nicoud J.-F., Kagan H. B., Tetrahedron Lett., 12, 2065–2068 (1971).
- 3) Taber D. F., Berry J. F., Martin T. J., J. Org. Chem., 73, 9334–9339 (2008).
- 4) Ford A., Miel H., Ring A., Slattery C. N., Maguire A. R., McKervey M. A., Chem. Rev., 115, 9981–10080 (2015).
- 5) Hashimoto S., Watanabe N., Anada M., Ikegami S., J. Synth. Org. Chem. Jpn. (Engl.), 54, 988–999 (1996).
- 6) Davies H. M. L., Beckwith R. E. J., Chem. Rev., 103, 2861–2904 (2003).
- 7) Doyle M. P., Duffy R., Ratnikov M., Zhou L., Chem. Rev., 110, 704–724 (2010).
- 8) García C. F., McKervey M. A., Ye T., Chem. Commun., 1465–1466 (1996).
- 9) Buck R. T., Moody C. J., Pepper A. G., ARKIVOC, viii, 16–33 (2002).
- 10) Saito H., Uchiyama T., Miyake M., Anada M., Hashimoto S., Takabatake T., Miyairi S., Heterocycles, 81, 1149–1155 (2010).
- 11) Xu B., Zhu S.-F., Xie X.-L., Shen J.-J., Zhou Q.-L., Angew. Chem. Int. Ed., 50, 11483–11486 (2011).
- 12) Xu B., Zhu S.-F., Zuo X.-D., Zhang Z.-C., Zhou Q.-L., Angew. Chem. Int. Ed., 53, 3913–3916 (2014).
- 13) Guo J.-X., Zhou T., Xu B., Zhu S.-F., Zhou Q.-L., Chem. Sci, 7, 1104–1108 (2016).
- 14) Dolling U. H., Davis P., Grabowski E. J. J., J. Am. Chem. Soc., 106, 446–447 (1984).
- 15) O’Donnell M. J., Bennett W. D., Wu S., J. Am. Chem. Soc., 111, 2353–2355 (1989).
- 16) Saito H., Miyairi S., Curr. Top. Med. Chem., 14, 224–228 (2014).
- 17) Saito H., Morita D., Uchiyama T., Miyake M., Miyairi S., Tetrahedron Lett., 53, 6662–6664 (2012).
- 18) Soai K., Yokoyama S., Hayasaka T., J. Org. Chem., 56, 4264–4268 (1991).
- 19) Soai K., Niwa S., Chem. Rev., 92, 833–856 (1992).
- 20) Saito H., Oishi H., Kitagaki S., Nakamura S., Anada M., Hashimoto S., Org. Lett., 4, 3887–3890 (2002).
- 21) Minami K., Saito H., Tsutsui H., Nambu H., Anada M., Hashimoto S., Adv. Synth. Catal., 347, 1483–1487 (2005).
- 22) Natori Y., Tsutsui H., Sato N., Nakamura S., Nambu H., Shiro M., Hashimoto S., J. Org. Chem., 74, 4418–4421 (2009).
- 23) Ito M., Namie R., Krishnamurthi J., Miyamae H., Takeda K., Nambu H., Hashimoto S., Synlett, 25, 288–292 (2014).
- 24) Ito M., Kondo Y., Nambu H., Anada M., Takeda K., Hashimoto S., Tetrahedron Lett., 56, 1397–1400 (2015).
- 25) Mealy M. J., Luderer M. R., Bailey W. F., Sommer M. B., J. Org. Chem., 69, 6042–6049 (2004).