Experimental
Chemistry1H-NMR and 13C-NMR spectra were recorded on Bruker AVANCE 400 spectrometer. Chemical shifts of protons are reported in parts per million downfield from tetramethylsilane. Peaks are labeled as single (s), doublet (d), triplet (t), double doublet (dd), doublet of triplets (dt), multiplet (m). The high-resolution mass spectra were analyzed on a SHIMADZU LCMS-IT-TOF mass spectrometer. All chemicals were purchased from Sigma-Aldrich, Energy and Alfa Aesar Chemical Companies and were used without further purification.
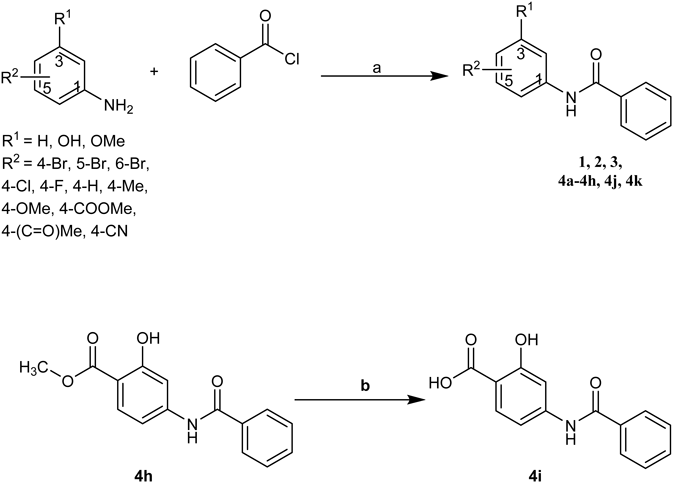
Typical Procedure for the Synthesis of Compounds 1–3, 4a–kA solution of BBr3 in dichloromethane (1.0 M, 12 mL, 12 mmol) was added slowly to a solution of 4-bromo-3-methoxyaniline (800 mg, 3.96 mmol) in methylene chloride (15 mL) at 0°C. The resulting brown solution was warmed to room temperature and stirred for 24 h. After saturated aqueous NaHCO3 (30 mL) was added at 0°C, the solution was extracted with EtOAc (20 mL × 3). The combined organic layer was dried with anhydrous Na2SO4, filtered and concentrated in vacuum. The residue was purified by flash chromatography over silica gel (petroleum–EtOAc = 2 : 1) to give 5-amino-2-bromophenol (665 mg, 88%).
To a solution of 5-amino-2-bromophenol (55 mg, 0.29 mmol) and triethylamine (53 µL, 0.38 mmol) in tetrahydrofuran (THF) (3 mL) was added slowly benzoyl chloride (0.32 mmol) at 0°C. The reaction mixture was then stirred at room temperature for 30 min. After the reaction was quenched with water (10 mL), the solution was extracted with EtOAc (10 mL × 2). The combined organic layer was dried over anhydrous Na2SO4, filtered and concentrated in vacuum. The residue was purified by column chromatography to afford the product 3 (74 mg, 87%).
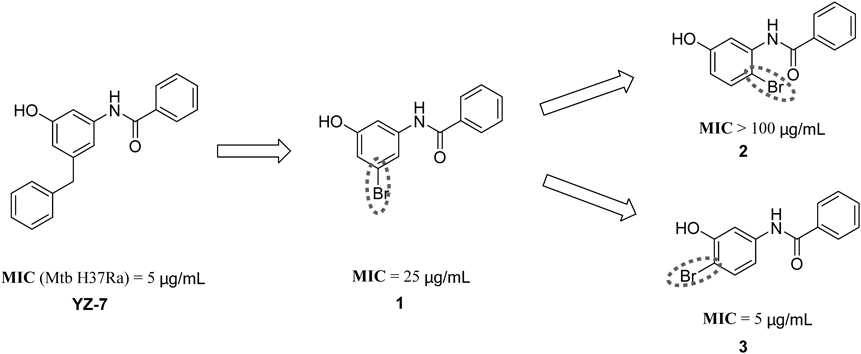
N-(4-Bromo-3-hydroxyphenyl)benzamide (3)White solid, mp 178.7–180.2°C; 1H-NMR (400 MHz, DMSO-d6) δ: 10.29 (s, 1H), 10.25 (s, 1H), 7.98–7.87 (m, 2H), 7.66 (d, J = 2.4 Hz, 1H), 7.61–7.56 (m, 1H), 7.52 (t, J = 7.3 Hz, 2H), 7.41 (d, J = 8.7 Hz, 1H), 7.12 (dd, J = 8.7, 2.4 Hz, 1H); 13C-NMR (101 MHz, dimethyl sulfoxide (DMSO)-d6) δ: 166.15, 154.42, 140.04, 135.33, 132.87, 132.11, 128.86, 128.15, 113.11, 108.68, 103.73; electrospray ionization (ESI)-MS m/z: 291.9944 (Calcd for C13H10NO2Br [M + H]+: 291.9968).
N-(3-Bromo-5-hydroxyphenyl)benzamide (1)The compound was synthesized via a similar procedure starting from 3-bromo-5-methoxyaniline. White solid, yield 80%. mp 184.3–186.2°C; 1H-NMR (400 MHz, DMSO-d6) δ: 10.25 (s, 1H), 9.96 (s, 1H), 7.94–7.89 (m, 2H), 7.62–7.57 (m, 1H), 7.55–7.50 (m, 2H), 7.49 (t, J = 1.8 Hz, 1H), 7.33 (t, J = 2.0 Hz, 1H), 6.68 (t, J = 2.0 Hz, 1H); 13C-NMR (101 MHz, DMSO-d6) δ: 166.24, 159.08, 141.93, 135.15, 132.23, 128.90, 128.16, 121.91, 113.96, 106.67; ESI-MS m/z: 291.9952 (Calcd for C13H10NO2Br [M + H]+: 291.9968).
N-(2-Bromo-5-hydroxyphenyl)benzamide (2)The compound was synthesized via a similar procedure starting from 2-bromo-5-methoxyaniline. White solid, yield 52%. mp 195.5–197.2°C; 1H-NMR (400 MHz, DMSO-d6) δ: 9.86 (d, J = 27.5 Hz, 2H), 7.97 (d, J = 6.9 Hz, 2H), 7.62–7.58 (m, 1H), 7.53 (dd, J = 10.8, 3.8 Hz, 2H), 7.45 (dd, J = 8.7, 2.9 Hz, 1H), 7.07 (s, 1H), 6.72–6.57 (m, 1H); 13C-NMR (101 MHz, DMSO-d6) δ: 165.32, 157.20, 136.99, 134.16, 132.91, 131.97, 128.65, 127.70, 115.26, 115.22, 108.45; ESI-MS m/z: 291.9922 (Calcd for C13H10NO2Br [M + H]+: 291.9968).
N-(4-Bromophenyl)benzamide (4a)The compound was synthesized via a similar procedure starting from 4-bromoaniline. White solid, yield 85%. mp 209.9–212.0°C; 1H-NMR (400 MHz, DMSO-d6) δ: 10.38 (s, 1H), 7.95 (d, J = 7.2 Hz, 2H), 7.77 (d, J = 8.8 Hz, 2H), 7.66–7.44 (m, 5H); 13C-NMR (101 MHz, DMSO-d6) δ: 165.69, 138.60, 134.72, 131.75, 131.46, 128.45, 127.70, 122.21, 115.34; ESI-MS m/z: 297.9822 (Calcd for C13H10NOBr [M + Na]+: 297.9838).
N-(4-Bromo-3-methoxyphenyl)benzamide (4b)The compound was synthesized via a similar procedure starting from 4-bromo-3-methoxyaniline. White solid, yield 89%. mp 134.6–136.5°C; 1H-NMR (400 MHz, DMSO-d6) δ: 10.35 (s, 1H), 7.96 (d, J = 7.3 Hz, 2H), 7.67 (d, J = 10.1 Hz, 1H), 7.63–7.48 (m, 4H), 7.40 (dd, J = 8.6, 2.1 Hz, 1H), 3.85 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ: 166.13, 155.75, 140.57, 135.18, 133.03, 132.20, 128.89, 128.11, 113.98, 105.26, 104.75, 56.45; ESI-MS m/z: 306.0110 (Calcd for C14H12NO2Br [M + H]+: 306.0124).
N-(4-Chloro-3-hydroxyphenyl)benzamide (4c)The compound was synthesized via a similar procedure starting from 5-amino-2-chlorophenol. White solid, yield 83%. mp 183.6–185.4°C; 1H-NMR (400 MHz, DMSO-d6) δ: 10.24 (d, J = 8.7 Hz, 2H), 7.92 (d, J = 7.2 Hz, 2H), 7.66 (d, J = 2.2 Hz, 1H), 7.56 (dt, J = 26.6, 7.2 Hz, 3H), 7.27 (d, J = 8.7 Hz, 1H), 7.17 (dd, J = 8.7, 2.2 Hz, 1H); 13C-NMR (101 MHz, DMSO-d6) δ: 166.09, 153.37, 139.39, 135.36, 132.07, 129.92, 128.84, 128.15, 114.59, 112.53, 108.88; ESI-MS m/z: 248.0462 (Calcd for C13H10NO2Cl [M + H]+: 248.0473).
N-(4-Fluoro-3-hydroxyphenyl)benzamide (4d)The compound was synthesized via a similar procedure starting from 5-amino-2-fluorophenol. Pale yellow solid, yield 83%. mp 180.1–182.3°C; 1H-NMR (400 MHz, DMSO-d6) δ: 10.18 (s, 1H), 9.93 (s, 1H), 7.99–7.90 (m, 2H), 7.63–7.56 (m, 2H), 7.53 (t, J = 7.3 Hz, 2H), 7.13 (m, 2H); 13C-NMR (101 MHz, DMSO-d6) δ: 165.48, 147.45 (d, JCF = 237.4 Hz), 144.57 (d, JCF = 13.1 Hz), 135.67(d, JCF = 2.0 Hz), 135.03, 131.57, 128.42, 127.68, 115.70 (d, JCF = 20.2 Hz), 111.19 (d, JCF = 6.0 Hz), 110.09; ESI-MS m/z: 232.0748 (Calcd for C13H10NO2F [M + H]+: 232.0768).
N-(3-Hydroxy-4-methylphenyl)benzamide (4f)The compound was synthesized via a similar procedure starting from 5-amino-2-methylphenol. White solid, yield 81%. mp 213.4–216.0°C; 1H-NMR (400 MHz, DMSO-d6) δ: 10.07 (s, 1H), 9.36 (s, 1H), 7.96–7.89 (m, 2H), 7.60–7.48 (m, 3H), 7.43 (d, J = 1.8 Hz, 1H), 7.05 (dd, J = 8.1, 1.9 Hz, 1H), 6.99 (d, J = 8.2 Hz, 1H), 2.09 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ: 165.32, 155.22, 137.78, 135.26, 131.39, 130.18, 128.35, 127.66, 119.24, 111.08, 107.17, 15.64; ESI-MS m/z: 228.1086 (Calcd for C14H13NO2 [M + H]+: 228.1019).
N-(3-Hydroxy-4-methoxyphenyl)benzamide (4g)The compound was synthesized via a similar procedure starting from 5-amino-2-methoxyphenol. White solid, yield 80%. mp 176.5–178.5°C; 1H-NMR (400 MHz, DMSO-d6) δ: 10.02 (s, 1H), 9.07 (s, 1H), 7.99–7.87 (m, 1H), 7.60–7.47 (m, 1H), 7.35 (d, J = 2.4 Hz, 1H), 7.14 (dd, J = 8.7, 2.4 Hz, 1H), 6.88 (d, J = 8.7 Hz, 1H), 3.74 (s, 1H); 13C-NMR (101 MHz, DMSO-d6) δ: 165.09, 146.28, 144.14, 135.23, 132.73, 131.35, 128.36, 127.59, 112.26, 111.23, 108.92, 55.88; ESI-MS m/z: 244.0927 (Calcd for C14H13NO3 [M + H]+: 244.0968).
Methyl 4-Benzamido-2-hydroxybenzoate (4h)The compound was synthesized via a similar procedure starting from methyl 4-amino-2-hydroxybenzoate. White solid, yield 77%. mp 170.0–171.8°C; 1H-NMR (400 MHz, DMSO-d6) δ: 10.65 (s, 1H), 10.51 (s, 1H), 7.94 (dd, J = 7.4, 5.9 Hz, 2H), 7.77 (d, J = 8.8 Hz, 1H), 7.63–7.57 (m, 2H), 7.57–7.51 (m, 2H), 7.37 (dd, J = 8.8, 2.0 Hz, 1H), 3.88 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ: 169.09, 166.23, 161.11, 145.76, 134.57, 131.99, 130.63, 128.49, 127.87, 111.41, 107.77, 107.14, 52.29; ESI-MS m/z: 294.0727 (Calcd for C15H13NO4 [M + Na]+: 294.0737).
Synthesis of 4-Benzamido-2-hydroxybenzoic Acid (4i)To a solution of compound 4h (90 mg, 0.33 mmol) in THF (2 mL) was added aqueous NaOH (1.5 M, 8 mL). The mixture was stirred at 50°C for 2 h. After being cooled to room temperature, the solution was acidified with 2 M HCl to pH = 3–4. The precipitation was collected to give the compound 4h (62 mg, 73%). mp 263.1–264.7°C; 1H-NMR (400 MHz, DMSO-d6) δ: 10.48 (s, 1H), 8.05–7.89 (m, 2H), 7.76 (d, J = 8.7 Hz, 1H), 7.67–7.44 (m, 4H), 7.34 (dd, J = 8.7, 2.0 Hz, 1H); 13C-NMR (101 MHz, DMSO-d6) δ: 172.11, 166.64, 162.44, 146.08, 135.06, 132.40, 131.32, 128.93, 128.30, 111.59, 108.51, 107.38; ESI-MS m/z: 280.0563 (Calcd for C14H11NO4 [M + Na]+: 280.0580).
N-(4-Acetyl-3-hydroxyphenyl)benzamide (4j)The compound was synthesized via a similar procedure starting from 1-(4-amino-2-hydroxyphenyl)ethan-1-one. Pale yellow solid, yield 82%. mp 204.9–206.6°C; 1H-NMR (400 MHz, DMSO-d6) δ: 12.34 (s, 1H), 10.54 (s, 1H), 7.95 (d, J = 8.0 Hz, 2H), 7.90 (d, J = 8.8 Hz, 1H), 7.65–7.59 (m, 1H), 7.59–7.50 (m, 3H), 7.37 (d, J = 8.8 Hz, 1H), 2.60 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ: 203.27, 166.31, 162.34, 146.29, 134.50, 132.42, 132.07, 128.52, 127.90, 115.83, 111.06, 106.96, 26.96; ESI-MS m/z: 278.0792 (Calcd for C15H13NO3 [M + Na]+: 278.0788).
N-(4-Cyano-3-hydroxyphenyl)benzamide (4k)The compound was synthesized via a similar procedure starting from 4-amino-2-methoxy benzonitrile. Yellow brown solid, yield 67%. mp 151.3–153.6°C; 1H-NMR (400 MHz, DMSO-d6) δ: 8.19–8.10 (m, 2H), 7.78 (t, J = 7.4 Hz, 1H), 7.63 (t, J = 7.7 Hz, 2H), 7.48 (d, J = 9.1 Hz, 1H), 6.60–6.54 (m, 2H), 6.45 (s, 2H); 13C-NMR (101 MHz, DMSO-d6) δ: 164.22, 155.14, 154.15, 134.99, 134.50, 130.38, 129.65, 128.57, 117.47, 111.90, 107.27, 90.59; ESI-MS m/z: 261.0582 (Calcd for C14H10N2O2 [M + Na]+: 261.0634).
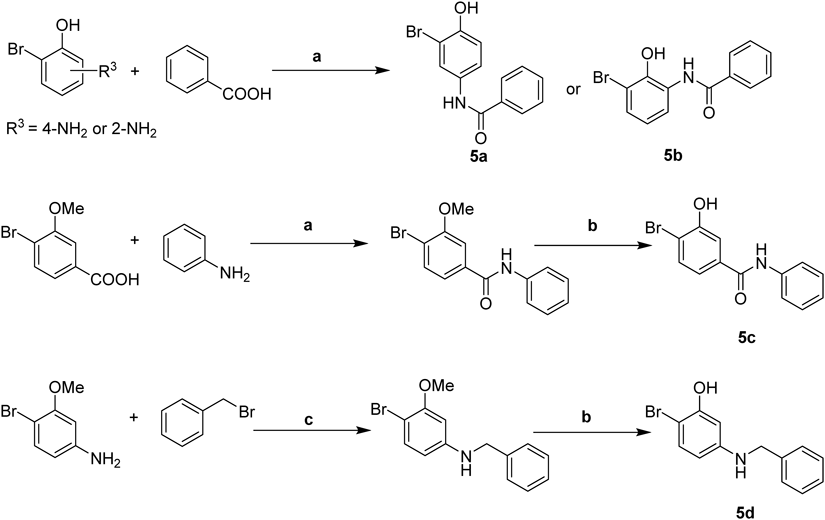
Synthesis of Compounds 5a–cTo a solution of benzoic acid (37 mg, 0.30 mmol) and HATU (138 mg, 0.36 mmol) in N,N-dimethylformamide (DMF) (4 mL) was added DIPEA (63 µL, 0.36 mmol) and 4-amino-2-bromophenol (56 mg, 0.30 mmol) separately. The reaction mixture was stirred at room temperature for 4 h. After water (20 mL) was added, the mixture was extracted with EtOAc (10 mL × 2). The combined organic layer was dried over anhydrous Na2SO4 and concentrated under vacuum. The residue was purified by flash chromatography over silica gel (petroleum–EtOAc = 10 : 1 to 5 : 1) to give the compound 5a.
N-(3-Bromo-4-hydroxyphenyl)benzamide (5a)White solid, yield 90%. mp 198.8–200.2°C; 1H-NMR (400 MHz, DMSO-d6) δ: 10.16 (s, 1H), 10.11 (s, 1H), 7.98 (d, J = 2.5 Hz, 1H), 7.92 (dd, J = 5.2, 3.3 Hz, 2H), 7.60–7.47 (m, 4H), 6.93 (d, J = 8.8 Hz, 1H); 13C-NMR (101 MHz, DMSO-d6) δ: 165.24, 150.44, 134.83, 131.86, 131.61, 128.48, 127.61, 124.95, 121.27, 116.03, 108.58; ESI-MS m/z: 291.9941 (Calcd for C13H10NO2Br [M + H]+: 291.9968).
N-(3-Bromo-2-hydroxyphenyl)benzamide (5b)The compound was synthesized via a similar procedure starting from 2-amino-6-bromophenol. White solid, yield 71%. mp 127.4–128.6°C; 1H-NMR (400 MHz, DMSO-d6) δ: 10.08 (s, 1H), 9.76 (s, 1H), 8.01 (d, J = 7.3 Hz, 2H), 7.62 (t, J = 7.3 Hz, 1H), 7.55 (t, J = 7.5 Hz, 2H), 7.44 (t, J = 6.7 Hz, 2H), 6.84 (t, J = 8.0 Hz, 1H); 13C-NMR (101 MHz, DMSO-d6) δ: 166.81, 148.04, 134.23, 132.43, 130.44, 128.91, 128.39, 127.91, 125.78, 121.05, 112.13; ESI-MS m/z: 291.9932 (Calcd for C13H10NO2Br [M + H]+: 291.9968).
4-Bromo-3-hydroxy-N-phenylbenzamide (5c)The compound was synthesized via a similar procedure starting from 4-bromo-3-hydroxybenzoic acid and aniline. White solid, yield 77%. mp 188.5–190.7°C; 1H-NMR (400 MHz, DMSO-d6) δ: 10.64 (s, 1H), 10.25 (s, 1H), 7.74 (d, J = 7.6 Hz, 2H), 7.64 (d, J = 8.2 Hz, 1H), 7.47 (d, J = 2.0 Hz, 1H), 7.38–7.30 (m, 3H), 7.10 (t, J = 7.4 Hz, 1H); 13C-NMR (101 MHz, DMSO-d6) δ: 164.85, 154.15, 139.05, 135.66, 132.81, 128.67, 123.79, 120.39, 119.36, 115.64, 113.05; ESI-MS m/z: 291.9819 (Calcd for C13H10NO2Br [M + H]+: 291.9968).
Synthesis of 5-(Benzylamino)-2-bromophenol (5d)To a solution of 4-bromo-3-methoxyaniline (80 mg, 0.40 mmol) and K2CO3 (110 mg, 0.80 mmol) in DMF (5 mL) was added (bromomethyl)benzene (75 mg, 0.44 mmol) at room temperature. The mixture was stirred at 55°C for 24 h. After water (20 mL) was added, the mixture was extracted with EtOAc (10 mL × 2). The combined organic layer was dried over anhydrous Na2SO4 and concentrated under vacuum. The crude product was purified by flash chromatography over silica gel (petroleum–EtOAc = 10 : 1 to 5 : 1) to give N-benzyl-4-bromo-3-methoxyaniline (84 mg, 72%).
The compound 5d was synthesized from N-benzyl-4-bromo-3-methoxyaniline via a procedure similar to “Typical Procedure for the Synthesis of Compounds 1–3, 4a–k.” White solid, yield 38%. mp 77.2–78.3°C; 1H-NMR (400 MHz, DMSO-d6) δ: 9.65 (s, 1H), 7.38–7.27 (m, 4H), 7.25–7.17 (m, 1H), 7.04 (d, J = 8.6 Hz, 1H), 6.36 (t, J = 5.9 Hz, 1H), 6.19 (d, J = 2.6 Hz, 1H), 6.02 (dd, J = 8.7, 2.6 Hz, 1H), 4.19 (d, J = 5.9 Hz, 2H); 13C-NMR (101 MHz, DMSO-d6) δ: 154.70, 149.67, 140.37, 132.88, 128.76, 127.54, 127.13, 106.19, 100.62, 95.37, 46.91; ESI-MS m/z: 278.0149 (Calcd for C13H12NOBr [M + H]+: 278.0175).
Synthesis of Compounds 6a–u. N-(4-Bromo-3-hydroxyphenyl)acetamide (6a)The compound was synthesized via a procedure similar to “Typical Procedure for the Synthesis of Compounds 1–3, 4a–k” using acetyl chloride. White solid, yield 54%. mp 215.7–216.5°C; 1H-NMR (400 MHz, DMSO-d6) δ: 10.22 (s, 1H), 9.93 (s, 1H), 7.47 (d, J = 2.4 Hz, 1H), 7.33 (d, J = 8.6 Hz, 1H), 6.94–6.79 (m, 1H), 2.01 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ: 168.41, 154.03, 139.74, 132.48, 111.25, 106.88, 102.39, 24.11; ESI-MS m/z: 229.9885 (Calcd for C8H8NO2Br [M + H]+: 229.9811).
N-(4-Bromo-3-hydroxyphenyl)acrylamide (6b)The compound was synthesized via a procedure similar to “Typical Procedure for the Synthesis of Compounds 1–3, 4a–k” using acryloyl chloride. Brown solid, yield 63%. mp 183.6–186.0°C; 1H-NMR (400 MHz, DMSO-d6) δ: 10.30 (s, 1H), 10.16 (s, 1H), 7.54 (d, J = 2.3 Hz, 1H), 7.38 (d, J = 8.6 Hz, 1H), 7.00 (dd, J = 8.7, 2.3 Hz, 1H), 6.43 (dd, J = 17.0, 10.1 Hz, 1H), 6.25 (dd, J = 17.0, 2.0 Hz, 1H), 5.76 (dd, J = 10.1, 2.0 Hz, 1H); 13C-NMR (101 MHz, DMSO-d6) δ: 163.12, 154.06, 139.37, 132.56, 131.76, 127.01, 111.64, 107.25, 103.04; ESI-MS m/z: 241.9805 (Calcd for C9H8NO2Br [M + H]+: 241.9811).
N-(4-Bromo-3-hydroxyphenyl)nonanamide (6c)The compound was synthesized via a similar procedure “Synthesis of Compounds 5a–c” using octanoic acid. White solid, yield 76%. mp 181.8–183.3°C; 1H-NMR (400 MHz, DMSO-d6) δ: 10.18 (s, 1H), 9.84 (s, 1H), 7.47 (d, J = 2.3 Hz, 1H), 7.32 (d, J = 8.6 Hz, 1H), 6.89 (dd, J = 8.7, 2.4 Hz, 1H), 2.26 (t, J = 7.4 Hz, 2H), 1.61–1.49 (m, 2H), 1.26 (dd, J = 5.7, 2.5 Hz, 8H), 0.85 (d, J = 7.0 Hz, 3H); 13C-NMR (101 MHz, DMSO-d6) δ: 171.36, 154.00, 139.74, 132.43, 111.33, 106.97, 102.31, 36.47, 31.19, 28.65, 28.48, 25.10, 22.09, 13.97; ESI-MS m/z: 314.0734 (Calcd for C14H20NO2Br [M + H]+: 314.0750).
N-(4-Bromo-3-hydroxyphenyl)cyclohexanecarboxamide (6d)The compound was synthesized via a similar procedure “Typical Procedure for the Synthesis of Compounds 1–3, 4a–k” using cyclohexanecarbonyl chloride. White solid, yield 80%. mp 194.9–197.2°C; 1H-NMR (400 MHz, DMSO-d6) δ: 10.19 (s, 1H), 9.79 (s, 1H), 7.46 (d, J = 2.3 Hz, 1H), 7.31 (d, J = 8.7 Hz, 1H), 6.90 (dd, J = 8.7, 2.4 Hz, 1H), 2.33–2.24 (m, 1H), 1.82–1.69 (m, 4H), 1.43–1.32 (m, 2H), 1.31–1.09 (m, 4H); 13C-NMR (101 MHz, DMSO-d6) δ: 174.44, 153.98, 139.90, 132.42, 111.49, 107.12, 102.33, 44.91, 29.13, 25.42, 25.25; ESI-MS m/z: 298.0391 (Calcd for C13H16NO2Br [M + H]+: 298.0437).
N-(4-Bromo-3-hydroxyphenyl)morpholine-4-carboxamide (6e)To a solution of 5-amino-2-bromophenol (55 mg, 0.29 mmol) and triethylamine (53 µL, 0.38 mmol) in THF (3 mL) was added slowly morpholine-4-carbonyl chloride (0.32 mmol) at 0°C. The mixture was stirred at 45°C for 12 h. The reaction was quenched with water (10 mL) and the mixture was extracted with EtOAc (10 mL × 2). The combined organic layer was dried over anhydrous Na2SO4, filtered and concentrated under vacuum. The residue was purified by column chromatography to afford the product 6e as a white solid (60 mg, 69%). mp 193.0–194.8°C; 1H-NMR (400 MHz, DMSO-d6) δ: 10.08 (s, 1H), 8.54 (s, 1H), 7.31 (d, J = 2.4 Hz, 1H), 7.27 (d, J = 8.7 Hz, 1H), 6.82 (dd, J = 8.7, 2.4 Hz, 1H), 3.62–3.56 (m, 4H), 3.42–3.38 (m, 4H); 13C-NMR (101 MHz, DMSO-d6) δ: 155.36, 154.22, 141.39, 132.48, 112.31, 107.88, 101.65, 66.44, 44.62; ESI-MS m/z: 301.0182 (Calcd for C11H13N2O3Br [M + H]+: 301.0143).
N-(4-Bromo-3-hydroxyphenyl)picolinamide (6f)The compound was synthesized via a similar procedure “Synthesis of Compounds 5a–c” using picolinic acid. White solid, yield 80%. mp 202.5–204.4°C; 1H-NMR (400 MHz, DMSO-d6) δ: 10.61 (s, 1H), 10.33 (s, 1H), 8.79–8.69 (m, 1H), 8.14 (d, J = 7.8 Hz, 1H), 8.10–8.02 (m, 1H), 7.80 (d, J = 2.4 Hz, 1H), 7.71–7.63 (m, 1H), 7.42 (d, J = 8.7 Hz, 1H), 7.21 (dd, J = 8.7, 2.4 Hz, 1H); 13C-NMR (101 MHz, DMSO-d6) δ: 162.57, 154.03, 149.84, 148.45, 138.74, 138.16, 132.48, 126.99, 122.46, 112.79, 108.18, 103.67; ESI-MS m/z: 292.9910 (Calcd for C12H9N2O2Br [M + H]+: 292.9920) .
N-(4-Bromo-3-hydroxyphenyl)thiophene-2-carboxamide (6g)The compound was synthesized via a similar procedure “Typical Procedure for the Synthesis of Compounds 1–3, 4a–k” using thiophene-2-carbonyl chloride. White solid, yield 66%. mp 222.7–224.5°C; 1H-NMR (400 MHz, DMSO-d6) δ: 10.31 (s, 1H), 10.21 (s, 1H), 8.01 (d, J = 3.1 Hz, 1H), 7.84 (d, J = 4.8 Hz, 1H), 7.57 (d, J = 2.2 Hz, 1H), 7.41 (d, J = 8.7 Hz, 1H), 7.24–7.16 (m, 1H), 7.09 (dd, J = 8.7, 2.2 Hz, 1H); 13C-NMR (101 MHz, DMSO-d6) δ: 159.88, 154.01, 139.91, 139.16, 132.50, 132.04, 129.29, 128.09, 112.60, 108.20, 103.40; ESI-MS m/z: 297.9494 (Calcd for C11H8NO2SBr [M + H]+: 297.9532).
N-(4-Bromo-3-hydroxyphenyl)furan-2-carboxamide (6h)The compound was synthesized via a similar procedure “Typical Procedure for the Synthesis of Compounds 1–3, 4a–k” using furan-2-carbonyl chloride. White solid, yield 53%. mp 207.2–209.7°C; 1H-NMR (400 MHz, DMSO-d6) δ: 10.32 (s, 1H), 10.19 (s, 1H), 7.93 (dd, J = 1.6, 0.7 Hz, 1H), 7.61 (d, J = 2.4 Hz, 1H), 7.43–7.31 (m, 2H), 7.09 (dd, J = 8.7, 2.4 Hz, 1H), 6.70 (dd, J = 3.5, 1.7 Hz, 1H); 13C-NMR (101 MHz, DMSO-d6) δ: 156.22, 154.00, 147.38, 145.90, 139.00, 132.49, 114.96, 112.66, 112.22, 108.21, 103.43; ESI-MS m/z: 281.9753 (Calcd for C11H8NO3Br [M + H]+: 281.9760).
N-(4-Bromo-3-hydroxyphenyl)-2-chlorobenzamide (6i)The compound was synthesized via a similar procedure “Typical Procedure for the Synthesis of Compounds 1–3, 4a–k” using 2-chlorobenzoyl chloride. White solid, yield 79%. mp 204.4–206.7°C; 1H-NMR (400 MHz, DMSO-d6) δ: 10.51 (s, 1H), 10.33 (s, 1H), 7.61 (d, J = 2.3 Hz, 1H), 7.56 (dd, J = 7.8, 2.5 Hz, 2H), 7.50 (m, J = 7.7, 1.7 Hz, 1H), 7.44 (m, J = 7.3, 1.1 Hz, 1H), 7.40 (d, J = 8.6 Hz, 1H), 7.01 (dd, J = 8.6, 2.3 Hz, 1H); 13C-NMR (101 MHz, DMSO-d6) δ: 165.02, 154.16, 139.30, 136.90, 132.66, 131.20, 129.91, 129.69, 128.95, 127.33, 111.93, 107.52, 103.43; ESI-MS m/z: 325.9586 (Calcd for C13H9NO2ClBr [M + H]+: 325.9578).
N-(4-Bromo-3-hydroxyphenyl)-3-chlorobenzamide (6j)The compound was synthesized via a similar procedure “Typical Procedure for the Synthesis of Compounds 1–3, 4a–k” using 3-chlorobenzoyl chloride. White solid, yield 74%. mp 184.2–186.1°C; 1H-NMR (400 MHz, DMSO-d6) δ: 10.35 (d, J = 3.6 Hz, 2H), 7.98 (t, J = 1.7 Hz, 1H), 7.89 (d, J = 7.8 Hz, 1H), 7.70–7.61 (m, 2H), 7.56 (t, J = 7.9 Hz, 1H), 7.42 (d, J = 8.7 Hz, 1H), 7.11 (dd, J = 8.7, 2.4 Hz, 1H); 13C-NMR (101 MHz, DMSO-d6) δ: 164.17, 154.03, 139.33, 136.86, 133.22, 132.50, 131.49, 130.44, 127.48, 126.58, 112.67, 108.24, 103.56; ESI-MS m/z: 325.9562 (Calcd for C13H9NO2ClBr [M + H]+: 325.9578).
N-(4-Bromo-3-hydroxyphenyl)-4-chlorobenzamide (6k)The compound was synthesized via the procedure similar to compound 6e. White solid, yield 52%. mp 195.0–197.3°C; 1H-NMR (400 MHz, DMSO-d6) δ: 10.32 (s, 1H), 10.19 (s, 1H), 7.93 (dd, J = 1.6, 0.7 Hz, 1H), 7.61 (d, J = 2.4 Hz, 1H), 7.43–7.31 (m, 2H), 7.09 (dd, J = 8.7, 2.4 Hz, 1H), 6.70 (dd, J = 3.5, 1.7 Hz, 1H); 13C-NMR (101 MHz, DMSO-d6) δ: 156.22, 154.00, 147.38, 145.90, 139.00, 132.49, 114.96, 112.66, 112.22, 108.21, 103.43; ESI-MS m/z: 325.9565 (Calcd for C13H9NO2ClBr [M + H]+: 325.9578).
N-(4-Bromo-3-hydroxyphenyl)-4-fluorobenzamide (6l)The compound was synthesized via the procedure similar to compound 3. White solid, yield 83%. mp 181.9–183.4°C; 1H-NMR (400 MHz, DMSO-d6) δ: 10.29 (d, J = 18.2 Hz, 2H), 8.11–7.95 (m, 2H), 7.64 (s, 1H), 7.38 (dt, J = 16.4, 8.7 Hz, 3H), 7.11 (d, J = 8.7 Hz, 1H); 13C-NMR (101 MHz, DMSO-d6) δ: 164.54, 164.12 (d, JCF = 249.5 Hz), 154.01, 139.52, 132.46, 131.32, 130.50 (d, JCF = 9.1 Hz), 115.36 (d, JCF = 22.2 Hz), 112.65, 108.22, 103.35; ESI-MS m/z: 309.9838 (Calcd for C13H9NO2FBr [M + H]+: 309.9873).
4-Bromo-N-(4-bromo-3-hydroxyphenyl)benzamide (6m)The compound was synthesized via a similar procedure “Typical Procedure for the Synthesis of Compounds 1–3, 4a–k” using 4-bromobenzoyl chloride. White solid, yield 81%. mp 208.5–210.1°C; 1H-NMR (400 MHz, DMSO-d6) δ: 10.31 (s, 2H), 7.93–7.81 (m, 2H), 7.80–7.68 (m, 2H), 7.63 (d, J = 2.4 Hz, 1H), 7.41 (d, J = 8.7 Hz, 1H), 7.10 (dd, J = 8.7, 2.4 Hz, 1H); 13C-NMR (101 MHz, DMSO-d6) δ: 164.66, 154.02, 139.41, 133.94, 132.48, 131.43, 129.88, 125.44, 112.65, 108.23, 103.47; ESI-MS m/z: 369.9088 (Calcd for C13H9NO2Br2 [M + H]+: 369.9073).
N-(4-Bromo-3-hydroxyphenyl)-4-methylbenzamide (6n)The compound was synthesized via a similar procedure “Typical Procedure for the Synthesis of Compounds 1–3, 4a–k” using 4-methylbenzoyl chloride. White solid, yield 62%. mp 195.4–197.2°C; 1H-NMR (400 MHz, DMSO-d6) δ: 10.30 (s, 1H), 10.17 (s, 1H), 7.85 (d, J = 8.2 Hz, 2H), 7.66 (d, J = 2.4 Hz, 1H), 7.40 (d, J = 8.7 Hz, 1H), 7.33 (d, J = 8.0 Hz, 2H), 7.11 (dd, J = 8.7, 2.4 Hz, 1H), 2.38 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ: 165.47, 153.98, 141.70, 139.71, 132.40, 132.01, 128.94, 127.78, 112.63, 108.20, 103.14, 21.07; ESI-MS m/z: 306.0111 (Calcd for C14H12NO2Br [M + H]+: 306.0124).
N-(4-Bromo-3-hydroxyphenyl)-4-methoxybenzamide (6o)The compound was synthesized via a similar procedure “Typical Procedure for the Synthesis of Compounds 1–3, 4a–k” using 4-methoxybenzoyl chloride. White solid, yield 81%. mp 207.6–208.9°C; 1H-NMR (400 MHz, DMSO-d6) δ: 10.26 (s, 1H), 10.09 (s, 1H), 7.93 (d, J = 8.8 Hz, 2H), 7.65 (d, J = 2.3 Hz, 1H), 7.39 (d, J = 8.7 Hz, 1H), 7.11 (dd, J = 8.7, 2.3 Hz, 1H), 7.06 (t, J = 5.8 Hz, 2H), 3.83 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ: 164.96, 161.95, 153.93, 139.80, 132.34, 129.67, 126.88, 113.60, 112.60, 108.16, 102.96, 55.46; ESI-MS m/z: 322.0041 (Calcd for C14H12NO3Br [M + H]+: 322.0073).
N-(4-Bromo-3-hydroxyphenyl)-4-(trifluoromethoxy)benzamide (6p)The compound was synthesized via a similar procedure “Typical Procedure for the Synthesis of Compounds 1–3, 4a–k” using 4-(trifluoromethoxy)benzoyl chloride. White solid, yield 80%. mp 189.1–190.1°C; 1H-NMR (400 MHz, DMSO-d6) δ: 10.35 (d, J = 10.6 Hz, 2H), 8.13–8.01 (m, 2H), 7.64 (d, J = 2.4 Hz, 1H), 7.52 (d, J = 8.0 Hz, 2H), 7.42 (d, J = 8.7 Hz, 1H), 7.11 (dd, J = 8.7, 2.4 Hz, 1H); 13C-NMR (101 MHz, DMSO-d6) δ: 164.49, 154.04, 139.41, 134.07, 132.51, 130.16, 120.75, 112.63, 108.21, 103.50, 13C-NMR (101 MHz, DMSO-d6) δ: 164.49, 154.04, 150.50, 139.41, 134.07, 132.51, 130.16, 120.75, 112.63, 108.21, 103.50; ESI-MS m/z: 375.9757 (Calcd for C14H9NO3F3Br [M + H]+: 375.9791).
N-(4-Bromo-3-hydroxyphenyl)-4-(dimethylamino)benzamide (6q)The compound was synthesized via a similar procedure “Typical Procedure for the Synthesis of Compounds 1–3, 4a–k” using 4-(dimethylamino)benzoyl chloride. White solid, yield 71%. mp 238.3–240.7°C; 1H-NMR (400 MHz, DMSO-d6) δ: 10.22 (s, 1H), 9.87 (s, 1H), 7.85 (d, J = 8.9 Hz, 2H), 7.67 (d, J = 2.3 Hz, 1H), 7.37 (d, J = 8.7 Hz, 1H), 7.11 (dd, J = 8.7, 2.3 Hz, 1H), 6.74 (d, J = 9.0 Hz, 2H), 2.99 (s, 6H); 13C-NMR (101 MHz, DMSO-d6) δ: 165.28, 153.89, 152.44, 140.19, 132.25, 130.96, 129.24, 120.93, 112.55, 110.76, 108.10, 102.52; ESI-MS m/z: 355.0345 (Calcd for C15H15N2O2Br [M + H]+: 335.0390).
N-(4-Bromo-3-hydroxyphenyl)-4-(trifluoromethyl)benzamide (6r)The compound was synthesized via a similar procedure “Typical Procedure for the Synthesis of Compounds 1–3, 4a–k” using 4-(trifluoromethyl)benzoyl chloride. White solid, yield 73%. mp 205.1–206.6°C; 1H-NMR (400 MHz, DMSO-d6) δ: 10.47 (s, 1H), 10.35 (s, 1H), 8.12 (d, J = 8.1 Hz, 2H), 7.90 (d, J = 8.3 Hz, 2H), 7.65 (d, J = 2.4 Hz, 1H), 7.43 (d, J = 8.7 Hz, 1H), 7.13 (dd, J = 8.7, 2.4 Hz, 1H); 13C-NMR (101 MHz, DMSO-d6) δ: 164.95, 154.50, 139.70, 139.16, 132.96, 131.85 (d, JCF = 32.3 Hz), 129.10, 125.83 (d, JCF = 3.0 Hz), 113.12, 108.72, 104.13; ESI-MS m/z: 359.9803 (Calcd for C14H9NO2F3Br [M + H]+: 359.9842).
N-(4-Bromo-3-hydroxyphenyl)-4-nitrobenzamide (6s)The compound was synthesized via a similar procedure “Typical Procedure for the Synthesis of Compounds 1–3, 4a–k” using 4-nitrobenzoyl chloride. Yellow solid, yield 48%. mp 237.0–239.1°C; 1H-NMR (400 MHz, DMSO-d6) δ: 10.58 (s, 1H), 10.38 (s, 1H), 8.37 (d, J = 8.8 Hz, 2H), 8.16 (d, J = 8.8 Hz, 2H), 7.65 (d, J = 2.2 Hz, 1H), 7.45 (d, J = 8.7 Hz, 1H), 7.14 (dd, J = 8.7, 2.3 Hz, 1H); 13C-NMR (101 MHz, DMSO-d6) δ: 164.45, 154.52, 149.62, 141.00, 139.58, 133.02, 129.74, 124.01, 113.11, 108.69, 104.28. ESI-MS m/z: 336.9884 (Calcd for C13H9N2O4Br [M + H]+: 336.9818).
N-(4-Bromo-3-hydroxyphenyl)-4-hydroxybenzamide (6t)The compound was synthesized by compound 6o as the material and then the procedure similar to “Typical Procedure for the Synthesis of Compounds 1–3, 4a–k.” White solid, yield 32%. mp 215.2–216.7°C; 1H-NMR (400 MHz, DMSO-d6) δ: 10.19 (d, J = 47.7 Hz, 2H), 9.99 (s, 1H), 7.82 (d, J = 8.3 Hz, 2H), 7.64 (s, 1H), 7.38 (d, J = 8.7 Hz, 1H), 7.09 (d, J = 8.7 Hz, 1H), 6.85 (d, J = 8.3 Hz, 2H); 13C-NMR (101 MHz, DMSO-d6) δ: 165.62, 161.06, 154.36, 140.37, 132.76, 130.25, 125.77, 115.36, 113.00, 108.56, 103.25; ESI-MS m/z: 307.9882 (Calcd for C13H10NO3Br [M + H]+: 307.9917).
Synthesis of N-(4-Bromo-3-hydroxyphenyl)-2-naphthamide (6u)The compound was synthesized via a similar procedure “Typical Procedure for the Synthesis of Compounds 1–3, 4a–k” using 2-naphthoyl chloride. White solid, yield 73%. mp 207.6–209.9°C; 1H-NMR (400 MHz, DMSO-d6) δ: 10.43 (s, 1H), 10.32 (s, 1H), 8.56 (s, 1H), 8.11–7.97 (m, 4H), 7.72 (d, J = 2.4 Hz, 1H), 7.68–7.58 (m, 2H), 7.43 (d, J = 8.7 Hz, 1H), 7.17 (dd, J = 8.7, 2.4 Hz, 1H); 13C-NMR (101 MHz, DMSO-d6) δ: 166.17, 154.48, 140.14, 134.75, 132.92, 132.67, 132.51, 129.44, 128.49, 128.34, 128.16, 127.35, 124.96, 113.11, 108.67, 103.76; ESI-MS m/z: 342.0097 (Calcd for C17H12NO2Br [M + H]+: 342.0124).
Biological AssaysEvaluation of Inhibitory Activity against Mtb H37Ra in VitroSelectable marker-free autoluminescent Mtb H37Ra (UAlRa)13) broth culture in Middlebook 7H9 medium plus 0.05% Tween80 and 10% oleic acid albumin dextrose catalase (OADC) (7H9-Tw-OADC). Upon reaching an OD600 of 0.5–0.8, relative light unit (RLU) value was determined by luminometer. When the RLU of 200 µL broth culture reached to over 1 million, the test compounds were prepare with two-fold serial dilutions in a range of final concentration from 100 to 0.0625 µg/mL per 200 µL UAlRa of broth culture (RLU were diluted to 2000–5000 per 200 µL). On the other hand, DMSO final concentration of 2% was used as negative control while the compounds (2-fold decreasing concentration from 100 to 0.0625 µg/mL) as well as isoniazid (0.1 µg/mL) and ethambutol (2 µg/mL) were used as positive controls. The RLU values were determined once daily until the 6th day. Data were analysed as MIC90 values which is basically the lowest drug concentration that achieved a RLUdrug/RLUDMSO of less than 10% after incubation.
Evaluation of Inhibitory Activity against Mtb H37Rv and MDR-TB StrainsThe MICs of the test compounds were determined by the microplate alamar blue assay (MABA)14) against Mtb H37Rv and clinically isolated MDR-TB strains isolated from the Guangzhou Chest Hospital. Mtb H37Rv and MDR-TB strains were cultured in 7H9-Tw-OADC. The test compounds were prepared with two-fold serial dilutions in a range of final concentration from 0.25 to 128 µg/mL, two-fold dilutions of compounds were prepared in 7H9-OADC medium in a 100 µL volume in 96-well microplates. INH, RIF and EMB were used as the positive controls. Bacterial suspension of inoculum of 2 × 105 CFU/mL was added 100 µL per well into the microplates, and the plates incubated 6 d at 37°C. On the 7th day, 12.5 µL of 20% Tween 80 and 20 µL of alamar Blue (Bio-Rad) were added to the test plate, and incubated another 24 h at 37°C, and then observed the color change. If a blue color in the well was deemed to no growth, and the blue color change to pink or violet was interpreted as growth. The MIC was defined as the lowest drug concentration which prevented a color change from blue to pink.
Determination of the Antibacterial ActivityTo evaluated compounds exclusive of against Mtb, we selected 6p and r for determination of antibacterial activity against Gram-positive and Gram-negative bacteria. Initially, the test compounds were dissolved in DMSO to prepare the stock solutions (10 mg/mL), and then prepared a range of concentrations from 50 to 0.4 µg/mL by means of the standard two fold serial dilution method in 96-well microtest plates. Amoxicillin was used as the positive controls. The bacterial suspension was adjusted with sterile saline to a concentration of 1 × 105 CFU. One hundred microliters the bacterial suspension per well was added into the plates and incubated at 37°C for 24 h. The MIC of compound was defined as the lowest concentration (the highest dilution) that completely inhibited the growth of bacteria after incubation at 37°C for 18–24 h.15)
Determination of the CytotoxicityA549 cell and HepG2 cell were used for determination of the cytotoxicity of compounds. This two cell lines both were culture in RPMI 1640 Medium (with 5% fetal bovine serum (FBS) and 1% penicillin–streptomycin), and incubated at 37°C in 5% CO2 until they reach log phage. The test compounds were diluted with RPMI 1640 Medium by 2-fold dilution and the concentrations from 100 to 3.125 µg/mL. The cells suspension (1 × 105 cell/mL) of 100 µL were seeded in 96-well plates and then incubated at 37°C in 5% CO2 overnight.16) One hundred microliters different concentrations of the test compound were added to the plate by the form of replace the cell medium and 100 µL RPMI 1640 Medium was used as the 0% inhibitor control. Each experiment was repeated three times. After being incubated for 48 h, 10 µL/well CCK8 was added in the plates, and another 1–2 h incubation, the plates were read in the Envision MultilabelReader at 450 nm. Cell survival rate (activity%) was calculated as follow: activity% = 100 × (ODcompound − ODblank)/(ODcontrol − ODblank). The cytotoxicities were reported as IC50 values, which were calculated by GraphPad Prism Software version 5.23.
Determination of the Liver Microsome StabilityThe rat liver microsome was purchased from Xenotech Company. The final incubation volume was set at 45 µL, contained reduced nicotinamide adenine dinucleotide phosphate (NADPH) (2 mM), microsomes (0.5 mg/mL) and the tested compound (1 µM). The incubation times were 0, 5, 15, 30, 45 and 60 min. Ice-cold acetonitrile was added to terminate the reaction and then centrifuged at 4°C for 15 min at 4000 rpm. The concentration of the tested compound in supernatant was determined by LC-MS.17)